

PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function
- PMID: 19435887
- PMCID: PMC2707195
- DOI: 10.1074/jbc.M109.008797
PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function
Abstract
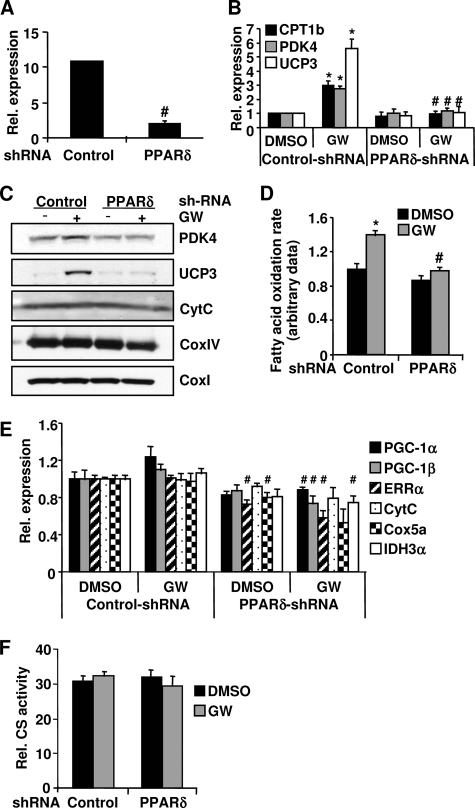
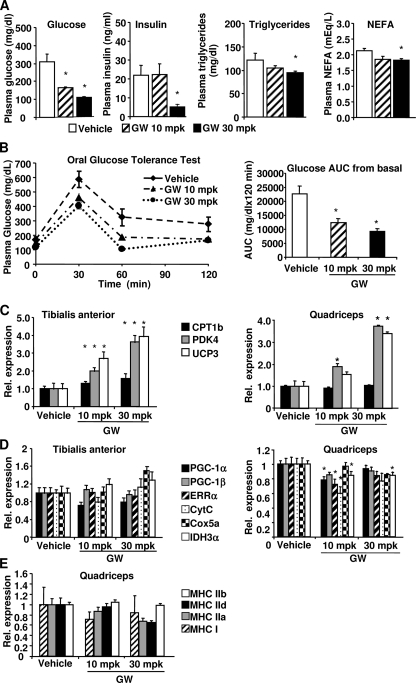
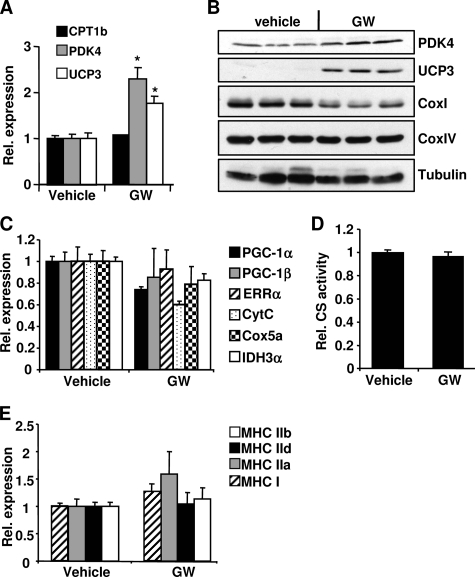
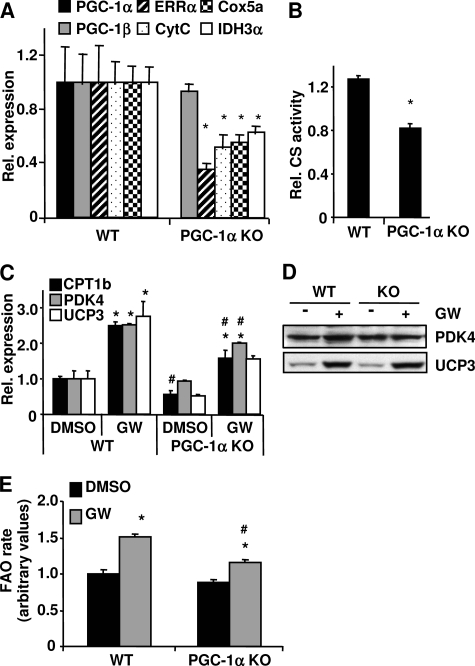
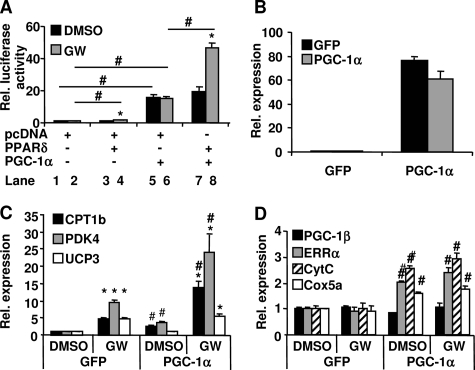
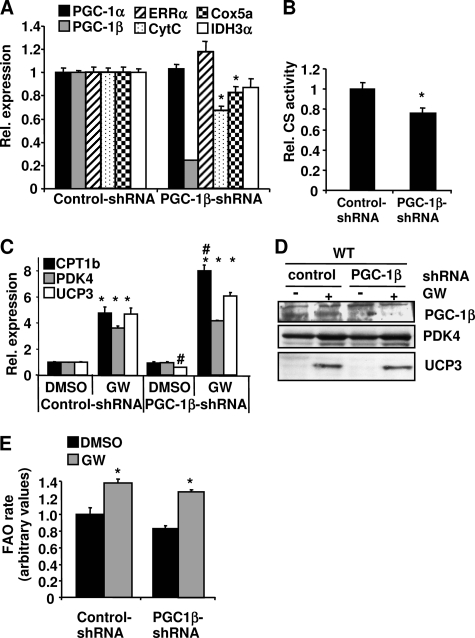
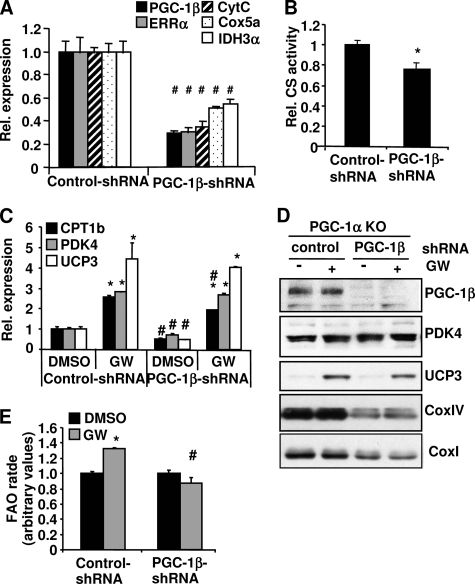
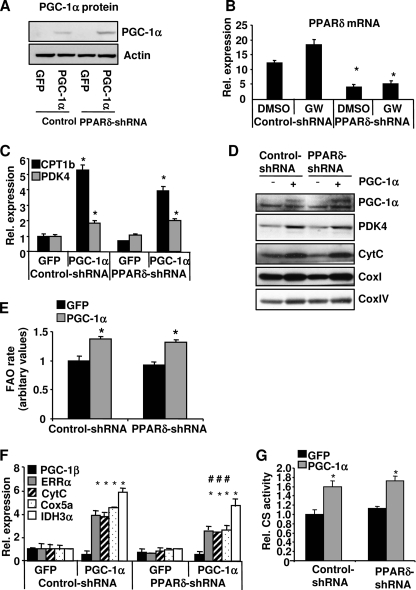
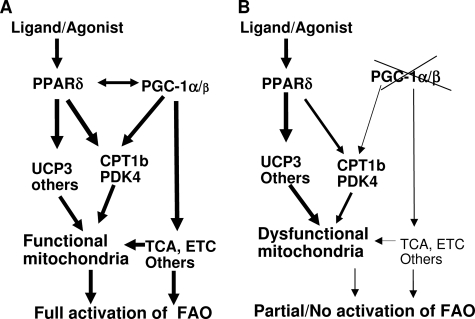
PPARdelta (peroxisome proliferator-activated receptor delta) is a regulator of lipid metabolism and has been shown to induce fatty acid oxidation (FAO). PPARdelta transgenic and knock-out mice indicate an involvement of PPARdelta in regulating mitochondrial biogenesis and oxidative capacity; however, the precise mechanisms by which PPARdelta regulates these pathways in skeletal muscle remain unclear. In this study, we determined the effect of selective PPARdelta agonism with the synthetic ligand, GW501516, on FAO and mitochondrial gene expression in vitro and in vivo. Our results show that activation of PPARdelta by GW501516 led to a robust increase in mRNA levels of key lipid metabolism genes. Mitochondrial gene expression and function were not induced under the same conditions. Additionally, the activation of Pdk4 transcription by PPARdelta was coactivated by PGC-1alpha. PGC-1alpha, but not PGC-1beta, was essential for full activation of Cpt-1b and Pdk4 gene expression via PPARdelta agonism. Furthermore, the induction of FAO by PPARdelta agonism was completely abolished in the absence of both PGC-1alpha and PGC-1beta. Conversely, PGC-1alpha-driven FAO was independent of PPARdelta. Neither GW501516 treatment nor knockdown of PPARdelta affects PGC-1alpha-induced mitochondrial gene expression in primary myotubes. These results demonstrate that pharmacological activation of PPARdelta induces FAO via PGC-1alpha. However, PPARdelta agonism does not induce mitochondrial gene expression and function. PGC-1alpha-induced FAO and mitochondrial biogenesis appear to be independent of PPARdelta.
Figures
References
-
- Takahashi S., Tanaka T., Sakai J. (2007) Endocr. J. 54, 347–357 - PubMed
-
- Braissant O., Foufelle F., Scotto C., Dauça M., Wahli W. (1996) Endocrinology 137, 354–366 - PubMed
-
- Luquet S., Lopez-Soriano J., Holst D., Fredenrich A., Melki J., Rassoulzadegan M., Grimaldi P. A. (2003) FASEB J., 03–0269fje - PubMed
-
- Handschin C., Spiegelman B. M. (2006) Endocr. Rev. 27, 728–735 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
