

Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer
- PMID: 19435893
- PMCID: PMC2724871
- DOI: 10.1158/0008-5472.CAN-09-0042
Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer
Abstract
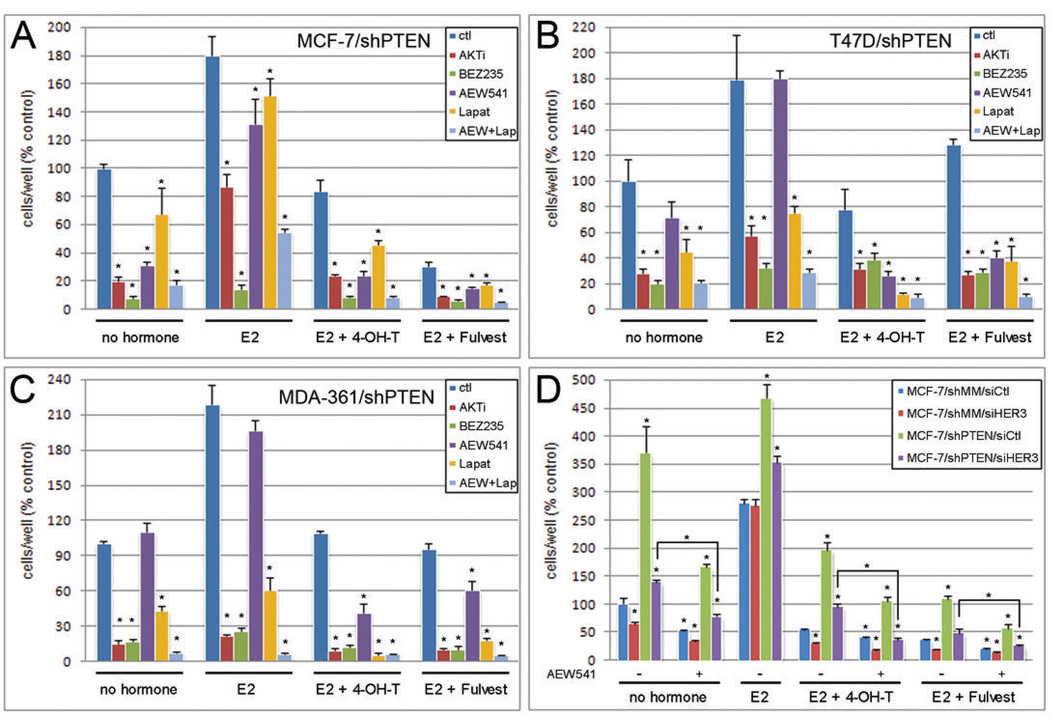
Knockdown of the tumor suppressor phosphatase Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) with shRNA in three estrogen receptor (ER)-positive breast cancer cell lines resulted in increased phosphatidylinositol-3 kinase (PI3K) and AKT activities, resistance to tamoxifen and fulvestrant, and hormone-independent growth. PTEN knockdown induced the up-regulation of ER transcriptional activity in MCF-7 cells but decreased ER protein levels and transcriptional activity in T47D and MDA-361 cells. Tamoxifen and fulvestrant treatment inhibited estradiol-induced ER transcriptional activity in all shPTEN cell lines but did not abrogate the increased cell proliferation induced by PTEN knockdown. PTEN knockdown increased basal and ligand-induced activation of the insulin-like growth factor-I (IGF-I) and ErbB3 receptor tyrosine kinases, and prolonged the association of the p85 PI3K subunit with the IGF-I receptor (IGF-IR) effector insulin receptor substrate-1 and with ErbB3, implicating PTEN in the modulation of signaling upstream of PI3K. Consistent with these data, PTEN levels inversely correlated with levels of tyrosine-phosphorylated IGF-IR in tissue lysate arrays of primary breast cancers. Inhibition of IGF-IR and/or ErbB2-mediated activation of ErbB3 with tyrosine kinase inhibitors restored hormone dependence and the growth inhibitory effect of tamoxifen and fulvestrant on shPTEN cells, suggesting that cotargeting both ER and receptor tyrosine kinase pathways holds promise for the treatment of patients with ER+, PTEN-deficient breast cancers.
Figures





References
-
- Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. - PubMed
-
- Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–2559. - PubMed
-
- Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol. 2001;14:672–676. - PubMed
-
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA080195/CA/NCI NIH HHS/United States
- K23 CA121994/CA/NCI NIH HHS/United States
- P50 CA98131/CA/NCI NIH HHS/United States
- P30 CA68485/CA/NCI NIH HHS/United States
- T32 CA078136/CA/NCI NIH HHS/United States
- K99 CA142899/CA/NCI NIH HHS/United States
- P50 CA098258/CA/NCI NIH HHS/United States
- R01 CA80195/CA/NCI NIH HHS/United States
- P50 CA098131/CA/NCI NIH HHS/United States
- R21 CA120248/CA/NCI NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- F32 CA121900/CA/NCI NIH HHS/United States
- T32 CA78136/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
