

Genomic disorders ten years on
- PMID: 19439022
- PMCID: PMC2684663
- DOI: 10.1186/gm42
Genomic disorders ten years on
Abstract
It is now becoming generally accepted that a significant amount of human genetic variation is due to structural changes of the genome rather than to base-pair changes in the DNA. As for base-pair changes, knowledge of gene and genome function has been informed by structural alterations that convey clinical phenotypes. Genomic disorders are a class of human conditions that result from structural changes of the human genome that convey traits or susceptibility to traits. The path to the delineation of genomic disorders is intertwined with the evolving technologies that have enabled the resolution of human genome analyses to continue increasing. Similarly, the ability to perform high-resolution human genome analysis has fueled the current and future clinical implementation of such discoveries in the evolving field of genome medicine.
Figures
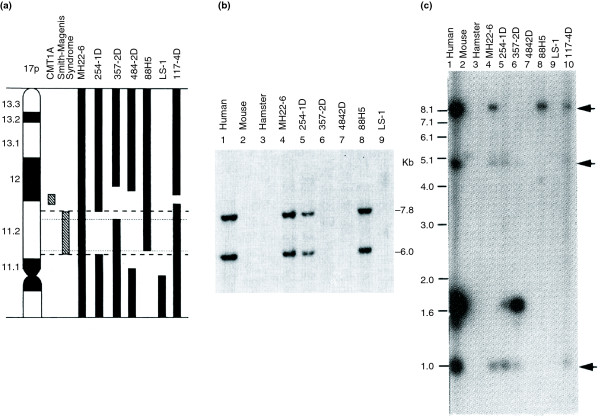
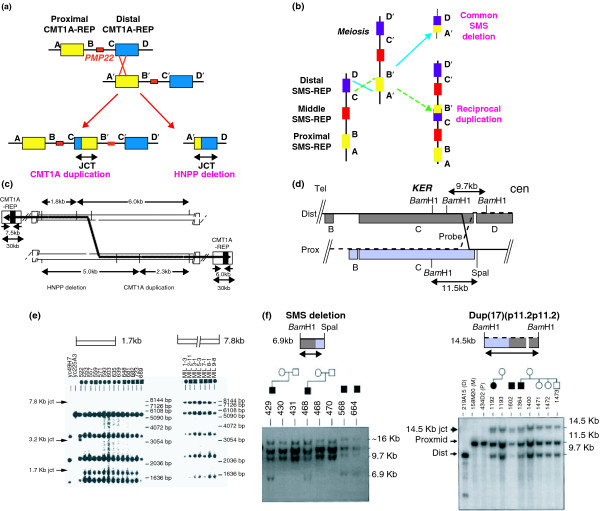
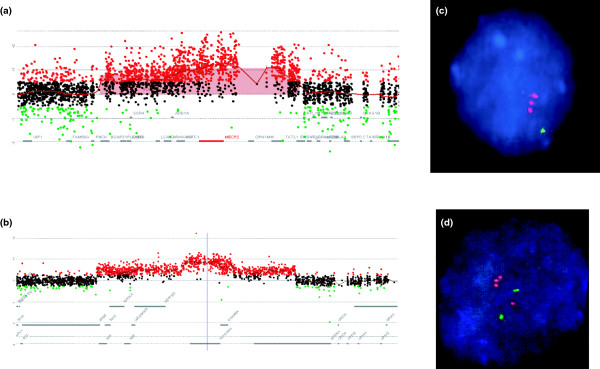
References
-
- Online Mendelian Inheritance in Man. http://hhttp://www.ncbi.nlm.nih.gov/sites/entrez?db=omim
-
- Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, Barker DF, Martin JJ, De Visser M, Bolhuis PA. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93–97. doi: 10.1016/0960-8966(91)90055-W. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
