

Prion protein biosynthesis and its emerging role in neurodegeneration
- PMID: 19447626
- PMCID: PMC3132587
- DOI: 10.1016/j.tibs.2009.03.001
Prion protein biosynthesis and its emerging role in neurodegeneration
Abstract
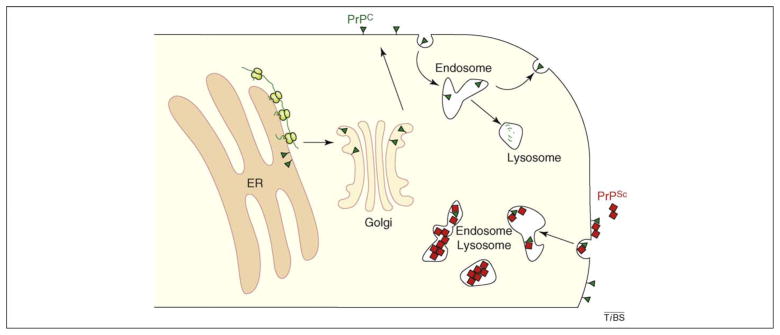
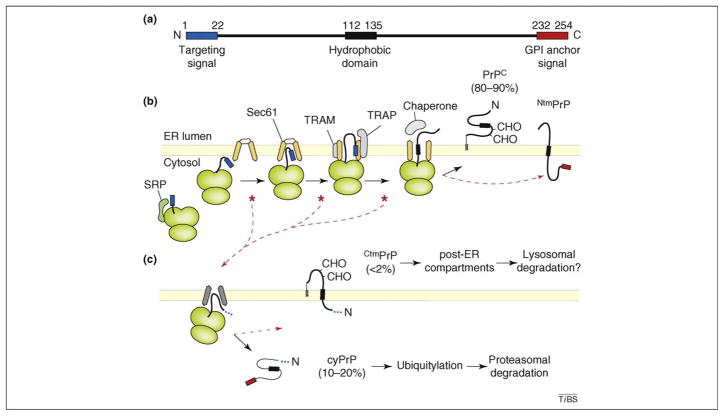
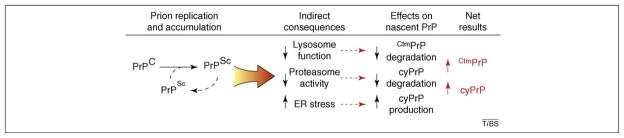
Various fatal neurodegenerative disorders are caused by altered metabolism of the prion protein (PrP). These diseases are typically transmissible by an unusual 'protein-only' mechanism in which a misfolded isomer, PrP(Sc), confers its aberrant conformation onto normal cellular PrP. An impressive range of studies has investigated nearly every aspect of this fascinating event; yet, our understanding of how PrP(Sc) accumulation might lead to cellular dysfunction and neurodegeneration is trifling. Recent advances in our understanding of normal PrP biosynthesis and degradation might have unexpectedly shed new light on this complex problem. Indeed, our current understanding of normal PrP cell biology, coupled with a growing appreciation of its complex metabolism, is providing new hypotheses for PrP-mediated neurodegeneration.
Figures
References
-
- Aguzzi A, et al. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 2008;3:11–40. - PubMed
-
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. - PubMed
-
- Prusiner SB, et al. Prion protein biology. Cell. 1998;93:337–348. - PubMed
-
- Wickner RB, et al. Prion genetics: new rules for a new kind of gene. Annu Rev Genet. 2004;38:681–707. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
