

Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes
- PMID: 19447966
- PMCID: PMC2704429
- DOI: 10.1101/gr.088633.108
Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes
Abstract
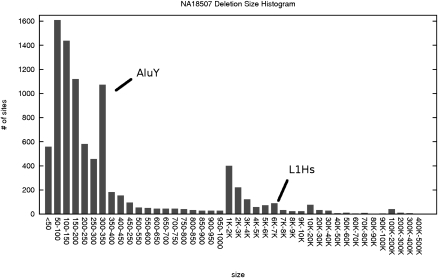
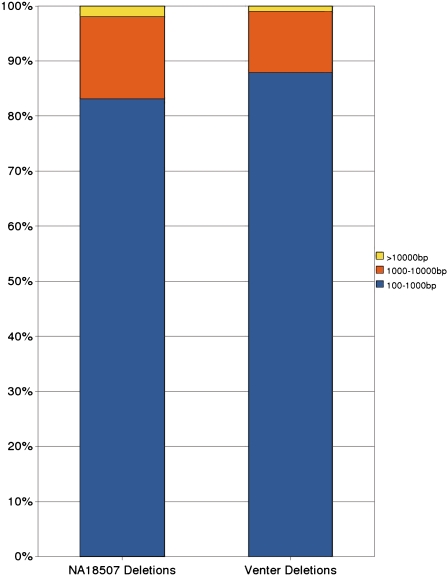
Recent studies show that along with single nucleotide polymorphisms and small indels, larger structural variants among human individuals are common. The Human Genome Structural Variation Project aims to identify and classify deletions, insertions, and inversions (>5 Kbp) in a small number of normal individuals with a fosmid-based paired-end sequencing approach using traditional sequencing technologies. The realization of new ultra-high-throughput sequencing platforms now makes it feasible to detect the full spectrum of genomic variation among many individual genomes, including cancer patients and others suffering from diseases of genomic origin. Unfortunately, existing algorithms for identifying structural variation (SV) among individuals have not been designed to handle the short read lengths and the errors implied by the "next-gen" sequencing (NGS) technologies. In this paper, we give combinatorial formulations for the SV detection between a reference genome sequence and a next-gen-based, paired-end, whole genome shotgun-sequenced individual. We describe efficient algorithms for each of the formulations we give, which all turn out to be fast and quite reliable; they are also applicable to all next-gen sequencing methods (Illumina, 454 Life Sciences [Roche], ABI SOLiD, etc.) and traditional capillary sequencing technology. We apply our algorithms to identify SV among individual genomes very recently sequenced by Illumina technology.
Figures
References
-
- Batzer M, Arcot S, Phinney J, Alegria-Hartman M, Kass D, Milligan S, Kimpton C, Gill P, Hochmeister M, Panayiotis A, et al. Genetic variation of recent Alu insertions in the human populations. J Mol Evol. 1996;42:22–29. - PubMed
-
- Boissinot S, Chevret P, Furano AV. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol. 2000;17:915–928. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources