

Identification of candidate regulatory SNPs by combination of transcription-factor-binding site prediction, SNP genotyping and haploChIP
- PMID: 19451166
- PMCID: PMC2709586
- DOI: 10.1093/nar/gkp381
Identification of candidate regulatory SNPs by combination of transcription-factor-binding site prediction, SNP genotyping and haploChIP
Abstract
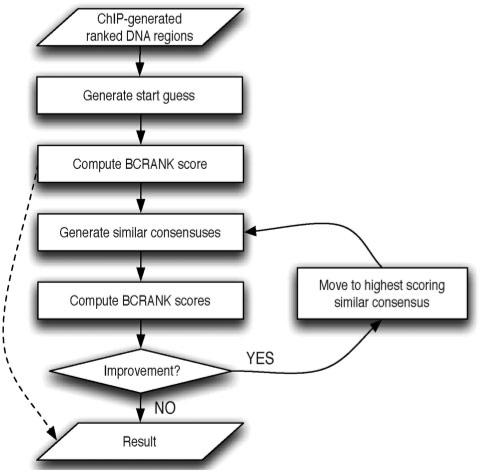
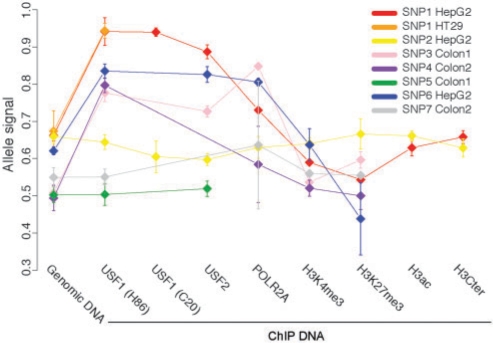
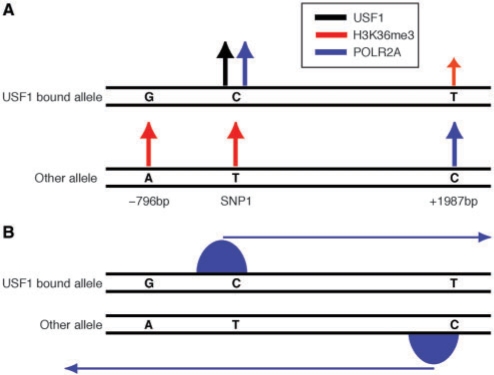
Disease-associated SNPs detected in large-scale association studies are frequently located in non-coding genomic regions, suggesting that they may be involved in transcriptional regulation. Here we describe a new strategy for detecting regulatory SNPs (rSNPs), by combining computational and experimental approaches. Whole genome ChIP-chip data for USF1 was analyzed using a novel motif finding algorithm called BCRANK. 1754 binding sites were identified and 140 candidate rSNPs were found in the predicted sites. For validating their regulatory function, seven SNPs found to be heterozygous in at least one of four human cell samples were investigated by ChIP and sequence analysis (haploChIP). In four of five cases where the SNP was predicted to affect binding, USF1 was preferentially bound to the allele containing the consensus motif. Allelic differences in binding for other proteins and histone marks further reinforced the SNPs regulatory potential. Moreover, for one of these SNPs, H3K36me3 and POLR2A levels at neighboring heterozygous SNPs indicated effects on transcription. Our strategy, which is entirely based on in vivo data for both the prediction and validation steps, can identify individual binding sites at base pair resolution and predict rSNPs. Overall, this approach can help to pinpoint the causative SNPs in complex disorders where the associated haplotypes are located in regulatory regions.
Availability: BCRANK is available from Bioconductor (http://www.bioconductor.org/).
Figures


References
-
- Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC, Taylor J, Burnett E, Gut I, Farrall M, et al. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. - PubMed
-
- Goring HH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, Cole SA, Jowett JB, Abraham LJ, Rainwater DL, Comuzzie AG, et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 2007;39:1208–1216. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
