

Pattern-based phylogenetic distance estimation and tree reconstruction
- PMID: 19455227
- PMCID: PMC2674673
Pattern-based phylogenetic distance estimation and tree reconstruction
Abstract
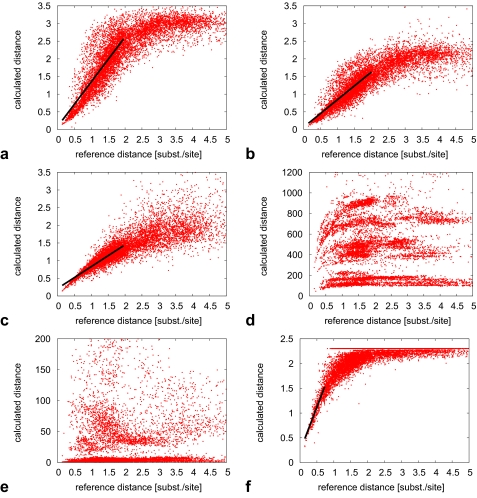
We have developed an alignment-free method that calculates phylogenetic distances using a maximum-likelihood approach for a model of sequence change on patterns that are discovered in unaligned sequences. To evaluate the phylogenetic accuracy of our method, and to conduct a comprehensive comparison of existing alignment-free methods (freely available as Python package decaf + py at http://www.bioinformatics.org.au), we have created a data set of reference trees covering a wide range of phylogenetic distances. Amino acid sequences were evolved along the trees and input to the tested methods; from their calculated distances we infered trees whose topologies we compared to the reference trees.We find our pattern-based method statistically superior to all other tested alignment-free methods. We also demonstrate the general advantage of alignment-free methods over an approach based on automated alignments when sequences violate the assumption of collinearity. Similarly, we compare methods on empirical data from an existing alignment benchmark set that we used to derive reference distances and trees. Our pattern-based approach yields distances that show a linear relationship to reference distances over a substantially longer range than other alignment-free methods. The pattern-based approach outperforms alignment-free methods and its phylogenetic accuracy is statistically indistinguishable from alignment-based distances.
Keywords: alignment-free methods; distance estimation; pattern discovery; phylogenetics.
Figures




Similar articles
-
Is multiple-sequence alignment required for accurate inference of phylogeny?Syst Biol. 2007 Apr;56(2):206-21. doi: 10.1080/10635150701294741. Syst Biol. 2007. PMID: 17454975 Free PMC article.
-
SATe-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees.Syst Biol. 2012 Jan;61(1):90-106. doi: 10.1093/sysbio/syr095. Epub 2011 Dec 1. Syst Biol. 2012. PMID: 22139466
-
PhyPA: Phylogenetic method with pairwise sequence alignment outperforms likelihood methods in phylogenetics involving highly diverged sequences.Mol Phylogenet Evol. 2016 Sep;102:331-43. doi: 10.1016/j.ympev.2016.07.001. Epub 2016 Jul 1. Mol Phylogenet Evol. 2016. PMID: 27377322
-
Phylogenetic Tree Estimation With and Without Alignment: New Distance Methods and Benchmarking.Syst Biol. 2017 Mar 1;66(2):218-231. doi: 10.1093/sysbio/syw074. Syst Biol. 2017. PMID: 27633353
-
SEQUENCE-FREE PHYLOGENETICS WITH MASS SPECTROMETRY.Mass Spectrom Rev. 2022 Jan;41(1):3-14. doi: 10.1002/mas.21658. Epub 2020 Nov 10. Mass Spectrom Rev. 2022. PMID: 33169385 Review.
Cited by
-
Is multiple-sequence alignment required for accurate inference of phylogeny?Syst Biol. 2007 Apr;56(2):206-21. doi: 10.1080/10635150701294741. Syst Biol. 2007. PMID: 17454975 Free PMC article.
-
Airborne microbial transport limitation to isolated Antarctic soil habitats.Nat Microbiol. 2019 Jun;4(6):925-932. doi: 10.1038/s41564-019-0370-4. Epub 2019 Mar 4. Nat Microbiol. 2019. PMID: 30833723
-
Prot-SpaM: fast alignment-free phylogeny reconstruction based on whole-proteome sequences.Gigascience. 2019 Mar 1;8(3):giy148. doi: 10.1093/gigascience/giy148. Gigascience. 2019. PMID: 30535314 Free PMC article.
-
An Information-Entropy Position-Weighted K-Mer Relative Measure for Whole Genome Phylogeny Reconstruction.Front Genet. 2021 Oct 22;12:766496. doi: 10.3389/fgene.2021.766496. eCollection 2021. Front Genet. 2021. PMID: 34745231 Free PMC article.
-
Spaced words and kmacs: fast alignment-free sequence comparison based on inexact word matches.Nucleic Acids Res. 2014 Jul;42(Web Server issue):W7-11. doi: 10.1093/nar/gku398. Epub 2014 May 14. Nucleic Acids Res. 2014. PMID: 24829447 Free PMC article.
References
-
- Apostolico A, Comin M, Parida L. Conservative extraction of overrepresented extensible motifs. In. Proceedings of the 13th International Conference on Intelligent Systems for Molecular Biology (ISMB 2005); 2005. pp. 223–233.
-
- Blaisdell B. Average values of a dissimilarity measure not requiring sequence alignment are twice the averages of conventional mismatch counts requiring sequence alignment for a computer-generated model system. . J. Mol. Evol. 1989;29(6):538–547. - PubMed
-
- Burstein D, Ulitsky I, Tuller T, Chor B. Information theoretic approaches to whole genome phylogenies. In. Proceedings of the Ninth Annual International Conference on Research in Computational Molecular Biology (RECOMB 2005); Cambridge, MA: 2005. pp. 283–295.
LinkOut - more resources
Full Text Sources