

Novel mutations affecting the Na, K ATPase alpha model complex neurological diseases and implicate the sodium pump in increased longevity
- PMID: 19455355
- PMCID: PMC2791699
- DOI: 10.1007/s00439-009-0673-2
Novel mutations affecting the Na, K ATPase alpha model complex neurological diseases and implicate the sodium pump in increased longevity
Abstract
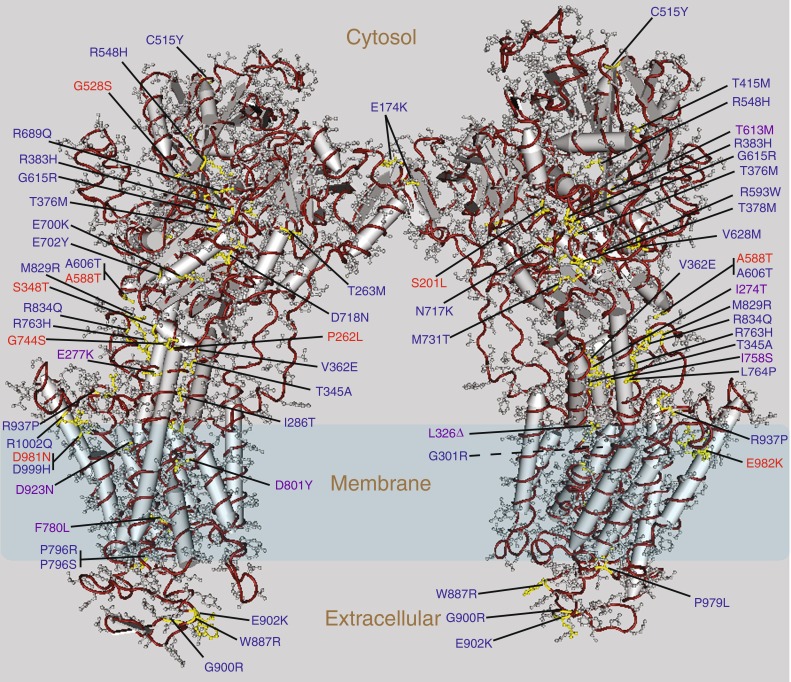
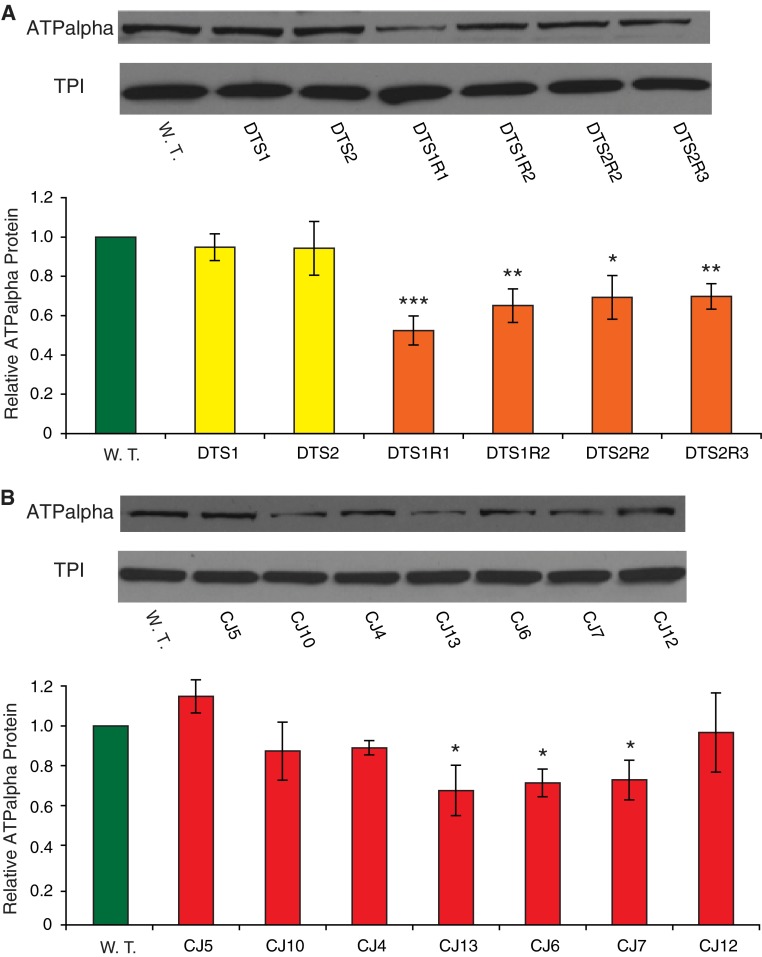
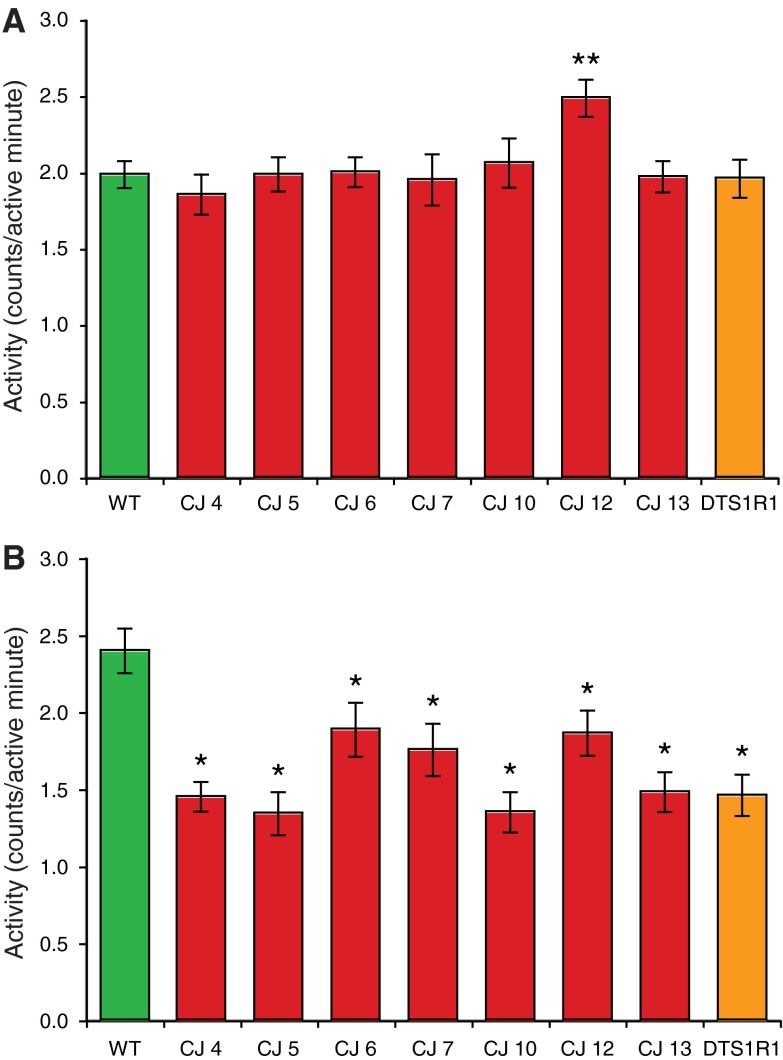
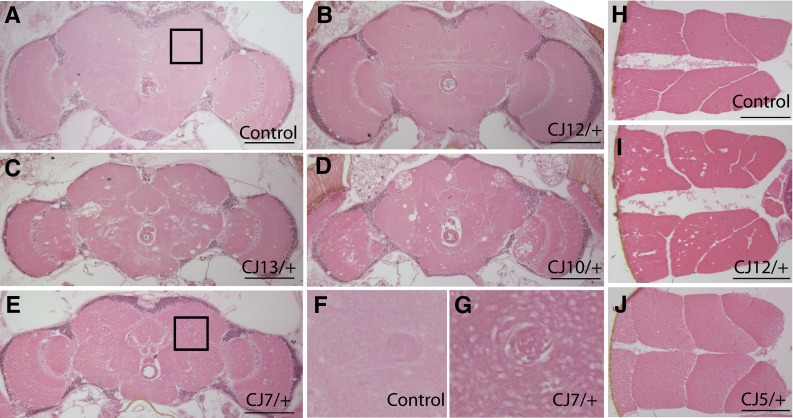
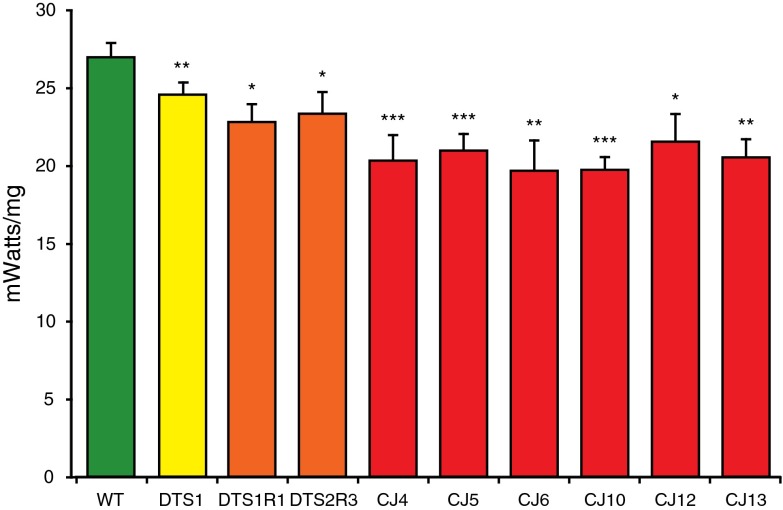
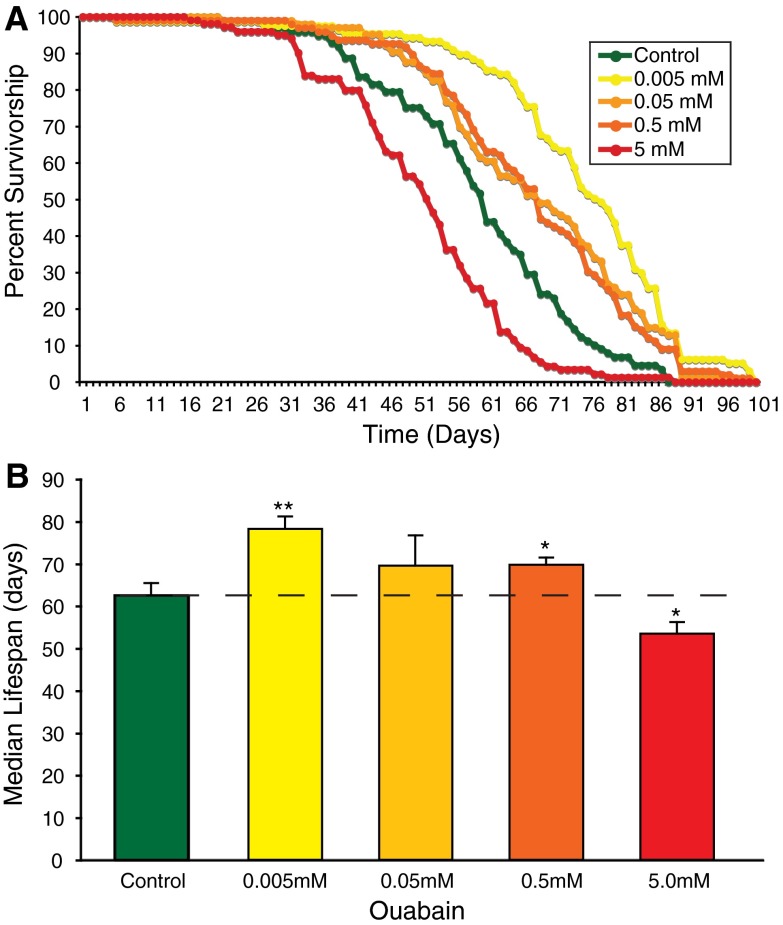
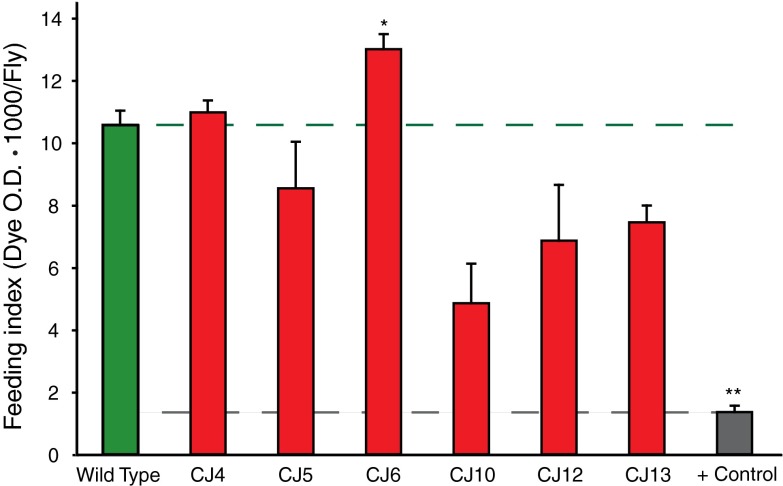
Mutations affecting the Na(+), K(+) ATPase alpha subunit have been implicated in at least two distinct human diseases, rapid-onset dystonia Parkinsonism (RDP), and familial hemiplegic migraine (FHM). Over 40 mutations have been mapped to the human ATP1A2 and ATP1A3 genes and are known to result in RDP, FHM or a variant of FHM with neurological complications. To develop a genetically tractable model system for investigating the role of the Na(+), K(+) ATPase in neural pathologies we performed genetic screens in Drosophila melanogaster to isolate loss-of-function alleles affecting the Na(+), K(+) ATPase alpha subunit. Flies heterozygous for these mutations all exhibit reduced respiration, consistent with a loss-of-function in the major ATPase. However, these mutations do not affect all functions of the Na(+), K(+) ATPase alpha protein since embryos homozygous for these mutations have normal septate junction paracellular barrier function and tracheal morphology. Importantly, all of these mutations cause neurological phenotypes and, akin to the mutations that cause RDP and FHM, these new alleles are missense mutations. All of these alleles exhibit progressive stress-induced locomotor impairment suggesting neuromuscular dysfunction, yet neurodegeneration is observed in an allele-specific manner. Surprisingly, studies of longevity demonstrate that mild hypomorphic mutations in the sodium pump significantly improve longevity, which was verified using the Na(+), K(+) ATPase antagonist ouabain. The isolation and characterization of a series of new missense alleles of ATPalpha in Drosophila provides the foundation for further studies of these neurological diseases and the role of sodium pump impairment in animal longevity.
Figures


References
-
- Ambrosini A, D’Onofrio M, Grieco GS, Di Mambro A, Montagna G, Fortini D, Nicoletti F, Nappi G, Sances G, Schoenen J, Buzzi MG, Santorelli FM, Pierelli F. Familial basilar migraine associated with a new mutation in the ATP1A2 gene. Neurology. 2005;65:1826–1828. doi: 10.1212/01.wnl.0000187072.71931.c0. - DOI - PubMed
-
- Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–F650. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
