

Transmembrane helix 12 modulates progression of the ATP catalytic cycle in ABCB1
- PMID: 19456124
- PMCID: PMC2791873
- DOI: 10.1021/bi900373x
Transmembrane helix 12 modulates progression of the ATP catalytic cycle in ABCB1
Abstract
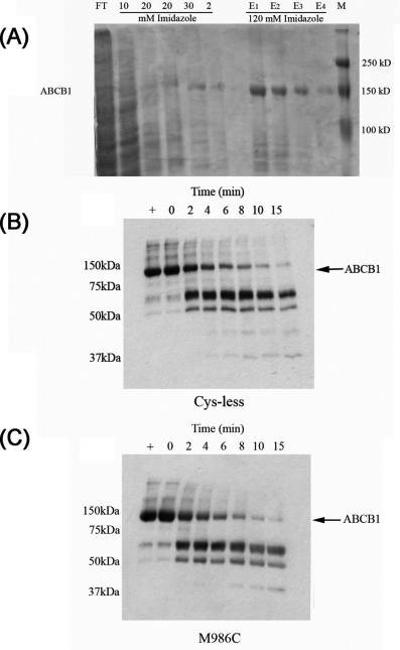
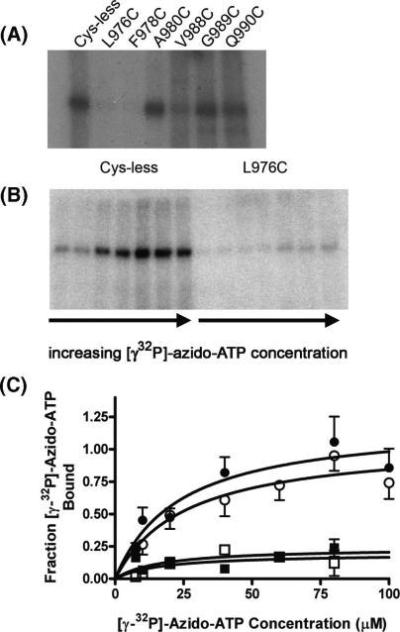
Multidrug efflux pumps, such as P-glycoprotein (ABCB1), present major barriers to the success of chemotherapy in a number of clinical settings. Molecular details of the multidrug efflux process by ABCB1 remain elusive, in particular, the interdomain communication associated with bioenergetic coupling. The present investigation has focused on the role of transmembrane helix 12 (TM12) in the multidrug efflux process of ABCB1. Cysteine residues were introduced at various positions within TM12, and their effect on ATPase activity, nucleotide binding, and drug interaction were assessed. Mutation of several residues within TM12 perturbed the maximal ATPase activity of ABCB1, and the underlying cause was a reduction in basal (i.e., drug-free) hydrolysis of the nucleotide. Two of the mutations (L976C and F978C) were found to reduce the binding of [gamma-(32)P]-azido-ATP to ABCB1. In contrast, the A980C mutation within TM12 enhanced the rate of ATP hydrolysis; once again, this was due to modified basal activity. Several residues also caused reductions in the potency of stimulation of ATP hydrolysis by nicardipine and vinblastine, although the effects were independent of changes in drug binding per se. Overall, the results indicate that TM12 plays a key role in the progression of the ATP hydrolytic cycle in ABCB1, even in the absence of the transported substrate.
Figures





References
-
- Pavelic ZP, Reising J, Pavelic L, Kelley DJ, Stambrook PJ, Gluckman JL. Detection of P-glycoprotein with four monoclonal antibodies in normal and tumor tissues. Arch. Otolaryngol., Head Neck Surg. 1993;119:753–757. - PubMed
-
- Smit JJ, Schinkel AH, Mol CA, Majoor D, Mooi WJ, Jongsma AP, Lincke CR, Borst P. Tissue distribution of the human MDR3 P-glycoprotein. Lab. Invest. 1994;71:638–649. - PubMed
-
- Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem. 1990;38:1277–1287. - PubMed
-
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
