

EDGAR: a software framework for the comparative analysis of prokaryotic genomes
- PMID: 19457249
- PMCID: PMC2696450
- DOI: 10.1186/1471-2105-10-154
EDGAR: a software framework for the comparative analysis of prokaryotic genomes
Abstract
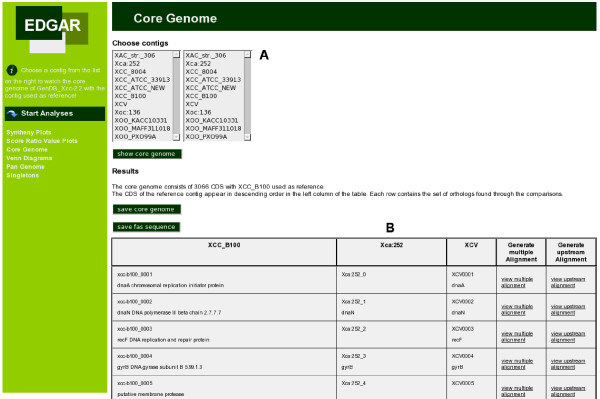
Background: The introduction of next generation sequencing approaches has caused a rapid increase in the number of completely sequenced genomes. As one result of this development, it is now feasible to analyze large groups of related genomes in a comparative approach. A main task in comparative genomics is the identification of orthologous genes in different genomes and the classification of genes as core genes or singletons.
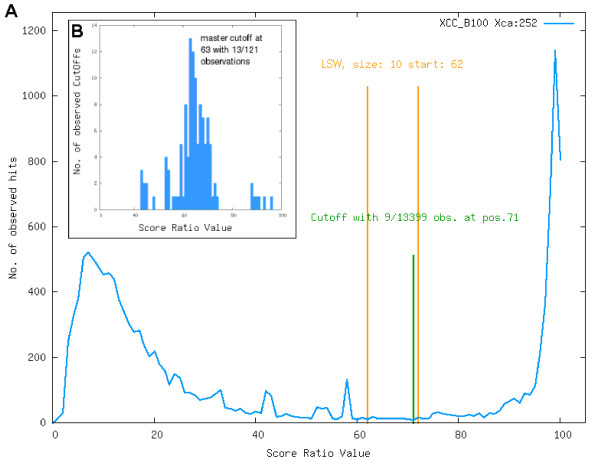
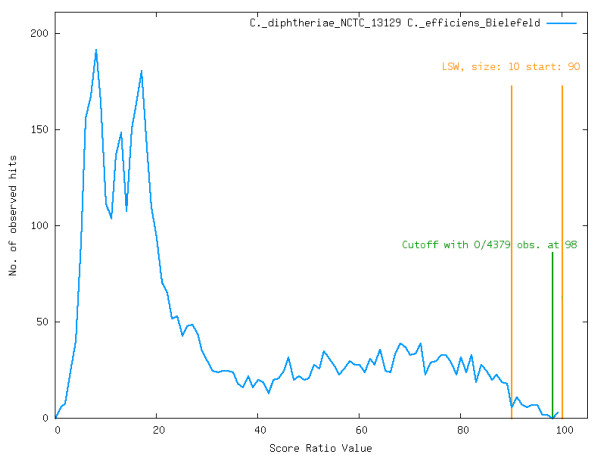
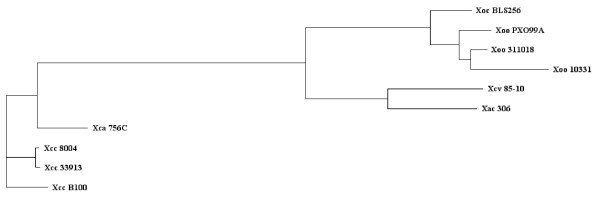
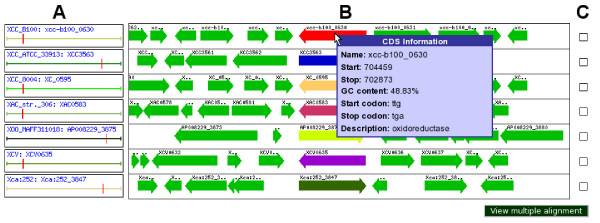
Results: To support these studies EDGAR - "Efficient Database framework for comparative Genome Analyses using BLAST score Ratios" - was developed. EDGAR is designed to automatically perform genome comparisons in a high throughput approach. Comparative analyses for 582 genomes across 75 genus groups taken from the NCBI genomes database were conducted with the software and the results were integrated into an underlying database. To demonstrate a specific application case, we analyzed ten genomes of the bacterial genus Xanthomonas, for which phylogenetic studies were awkward due to divergent taxonomic systems. The resultant phylogeny EDGAR provided was consistent with outcomes from traditional approaches performed recently and moreover, it was possible to root each strain with unprecedented accuracy.
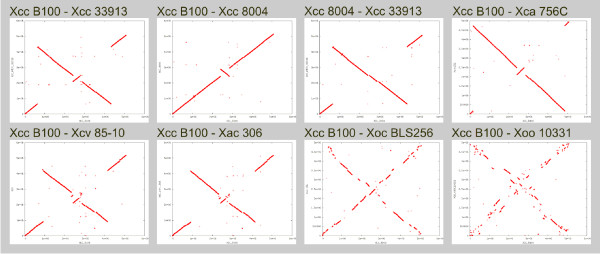
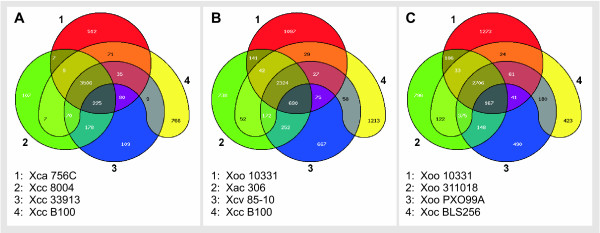
Conclusion: EDGAR provides novel analysis features and significantly simplifies the comparative analysis of related genomes. The software supports a quick survey of evolutionary relationships and simplifies the process of obtaining new biological insights into the differential gene content of kindred genomes. Visualization features, like synteny plots or Venn diagrams, are offered to the scientific community through a web-based and therefore platform independent user interface http://edgar.cebitec.uni-bielefeld.de, where the precomputed data sets can be browsed.
Figures
References
-
- Hollricher K. Microbial systematics – Species Don't Really Mean Anything in the Bacterial World. Lab Times. 2007;5:22–25.
-
- Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore LH, More WEC, Murray RGE, Stackebrandt E, Starr MP, Trüper HG. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. International Journal of Systematic Bacteriology. 1987;37:463–464.
-
- Hiller NL, Janto B, Hogg JS, Boissy R, Yu S, Powell E, Keefe R, Ehrlich NE, Shen K, Hayes J, Barbadora K, Klimke W, Dernovoy D, Tatusova T, Parkhill J, Bentley SD, Post JC, Ehrlich GD, Hu FZ. Comparative genomic analyses of seventeen Streptococcus pneumoniae strains: insights into the Pneumococcal Supragenome. Journal of Bacteriology. 2007;189:8186–8195. - PMC - PubMed
-
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, y Ros IM, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJB, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13950–13955. - PMC - PubMed
-
- Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Current Opinion in Genetics & Development. 2005;15:589–594. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
