

Review
doi: 10.1007/s11916-009-0034-9.
Contribution of primary afferent channels to neuropathic pain
Affiliations
- PMID: 19457280
- PMCID: PMC2859626
- DOI: 10.1007/s11916-009-0034-9
Item in Clipboard
Review
Contribution of primary afferent channels to neuropathic pain
Curr Pain Headache Rep.
2009 Jun.
Abstract
Neuropathic pain remains a serious medical problem because of patient morbidity and the absence of effective therapeutic interventions. Recent evidence suggests that this type of pain may be particularly difficult to manage because underlying mechanisms are influenced by a variety of factors, including type of injury, site of injury, and time after injury. This situation is exacerbated by the fact that different mechanisms may contribute to unique aspects of neuropathic pain, including ongoing pain as well as mechanical and thermal hypersensitivity. The different ion channels present in primary afferent neurons implicated in each of these aspects of neuropathic pain are reviewed.
Figures
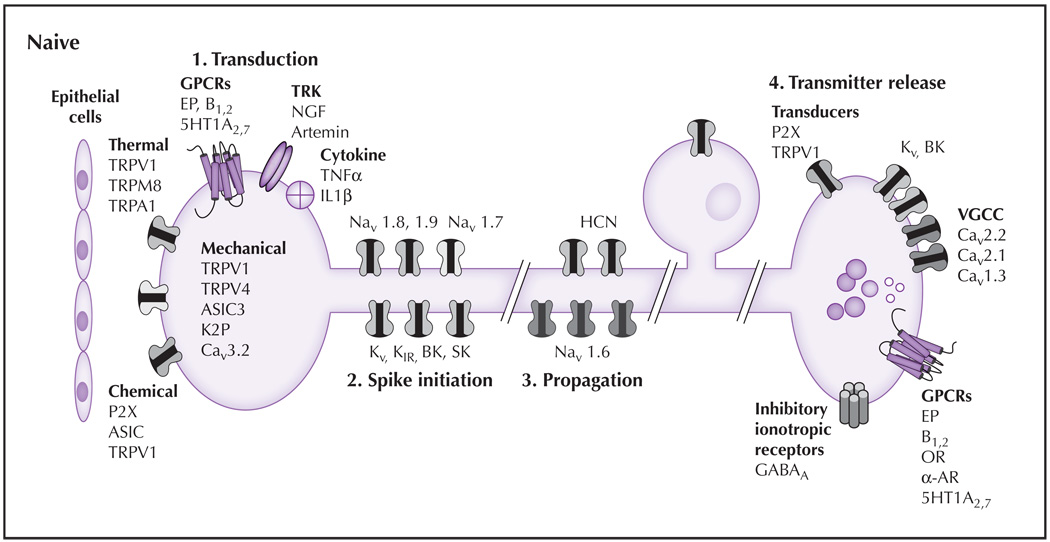
The principal sensory functions of nociceptive afferents consist of transduction, spike initiation, propagation, and transmitter release. (1) Transduction: In naive tissue, proteins thought to play a role in mechanotransduction include TRPV4, ASIC-3, and Cav3.2. Several different classes of TRP channels are involved in transduction of temperature from noxious cold (TRPA1), cool (TRPM8), warm (TRPV4), and hot (TRPV1). Ionotropic chemoreceptors present in nociceptive afferents include TRPV1, ASIC3, and P2X3. A wide variety of metabotropic receptors also are present on the terminals of nociceptive afferents including GPCRs, which are responsive to prostaglandins (EP, bradykinin, 5HT), TRK, and receptors for cytokines. (2) Spike initiation: Action potential threshold is regulated by several K+ channels (eg, Kv, KIR, K2P, BK, SK). HCN channels also contribute to action potential threshold. Nav1.9 also may contribute to establishing action potential threshold. The channels responsible for the upstroke of the action potential include Nav1.7 and Nav1.8. (3) Spike propagation: The ion channels underlying action potential propagation are distinct from those underlying spike initiation and include Nav1.6. HCN channels also contribute to spike propagation. (4) Transmitter release: The release of transmitter at the central terminals of nociceptive afferents is dependent on VGCCs, which are modulated following activation of inhibitory GPCRs and serve as the primary mechanism for the therapeutic efficacy of OR and α-AR agonists. Transmitters present in nociceptive afferents are generally packaged in small clear vesicles (which generally contain the excitatory amino acid glutamate) and large dense core vesicles (which contain, among other things, neuropeptides [eg, substance P, calcitonin generelated peptide]). A number of excitatory transducers (eg, P2X3 and TRPV1) appear to facilitate central transmitter release. Inhibition of the central terminal may involve activation of Kv, BK channels, and presynaptic GABAA receptors. Excitatory GPCRs (eg, EP, B1,2 receptors) also are present. 5HT—serotonin; AR—adrenergic receptor; ASIC—acid-sensing ion channel; B—bradykinin; BK—large conductance Ca2+ dependent channel; GABA—γ-aminobutyric acid; GPCR—G protein–coupled receptor; HCN—hyperpolarization-activated cyclic nucleotide channel; IL—interleukin; NGF—nerve growth factor; OR—opioid receptor; SK—small conductance Ca2+ dependent channel; TNF—tumor necrosis factor; TRK—tyrosine receptor kinases; TRPA—transient receptor potential ankyrin; TRPM—transient receptor potential melastatin; TRPV—transient receptor potential vanilloid; VGCC—voltage-dependent Ca2+ channel.
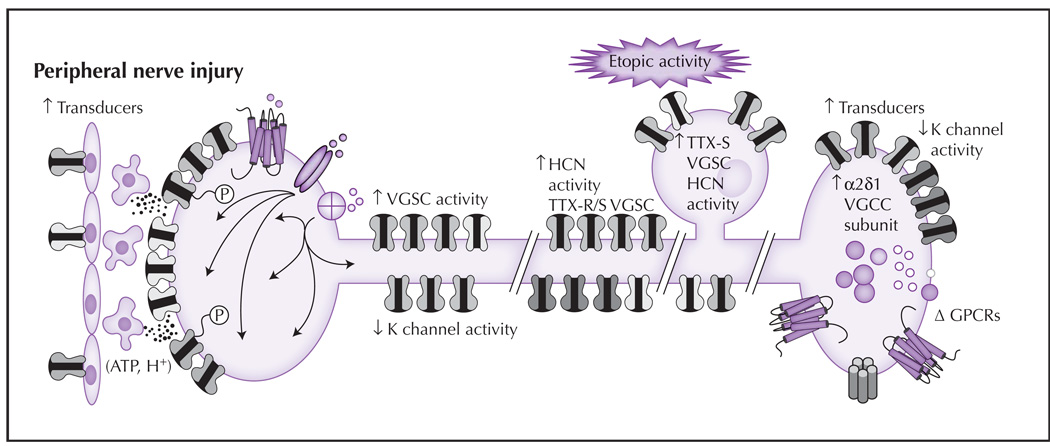
Following partial nerve injury, changes in nociceptive terminals of the spared or uninjured afferent result in an increase in sensitivity to noxious stimuli and the emergence of membrane depolarization. Similar changes also may occur at sites of damage in the injured afferents, although the mechanisms driving the changes appear to be different. (1) Transduction: There are increases in the density of several transducers and post-translational modifications (depicted as phosphorylation, P) that increase channel activity or sensitivity. These events are facilitated by release of mediators from epithelial cells. (2) Spike initiation: Changes following nerve injury include a decrease in K+ channel density and/or current, and an increase in HCN and Na+ channel density and/or activity. (3) Spike propagation: The pattern of channels influencing propagation can change following injury, including redistribution of Nav1.7 and/or Nav1.8 to the cell membrane. (4) Ectopic activity: In the presence of nerve injury, changes in the soma, sites of injury, and/or other sites along the axon, such as an increase in sodium and HCN channels, may lead to membrane instability (manifest as oscillatory behavior). The aberrant distribution of transducers also has been described. (5) Transmitter release: Changes that occur in the central terminals of nociceptive afferents after nerve injury include an increase in the α2δ1-subunit of VGCCs. This subunit is important for trafficking channels to the membrane. There also is an increase in neuropeptide expression, the emergence of additional excitatory receptors, and a decrease in K+ currents that facilitate nociceptive signaling. ATP—adenosine triphosphate; GPCR—G protein–coupled receptor; HCN—hyperpolarization-activated cyclic nucleotide channel; TTX—tetrodotoxin; VGCC—voltage-dependent Ca2+ channel; VGSC—voltage-gated Na+ channel.
References
-
- Gold MS, Chessell I, Devor M, et al. Peripheral nervous system targets: Rapporteur report. In: Campbell JN, Basbaum AI, Dray A, et al., editors. Emerging Strategies for the Treatment of Neuropathic Pain. Seattle: IASP Press; 2006. pp. 3–36.
-
- Backonja MM, Stacey B. Neuropathic pain symptoms relative to overall pain rating. J Pain. 2004;5:491–497. - PubMed
-
- Dubner R. The neurobiology of persistent pain and its clinical implications. Suppl Clin Neurophysiol. 2004;57:3–7. - PubMed
-
- Coull JA, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources