

Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development
- PMID: 19457928
- PMCID: PMC2714727
- DOI: 10.1093/hmg/ddp241
Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development
Abstract
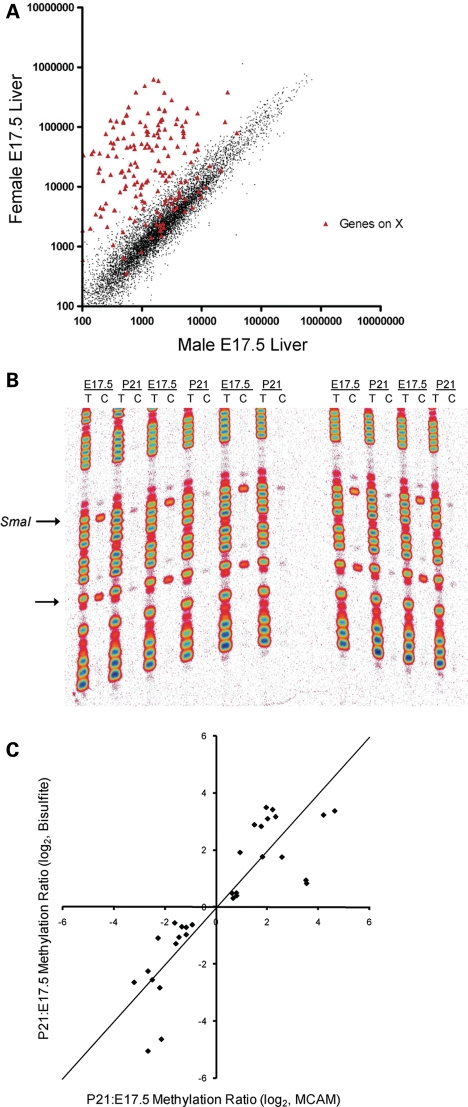
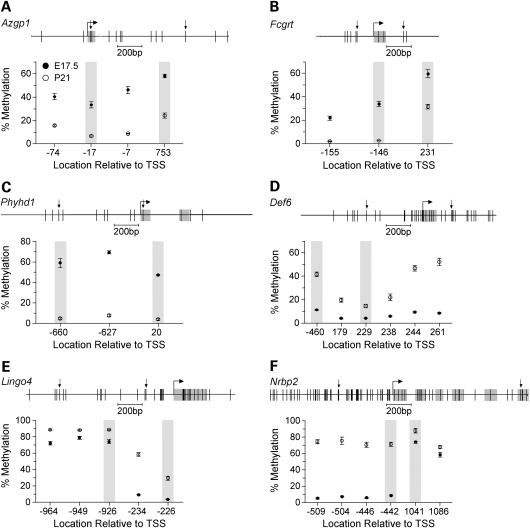
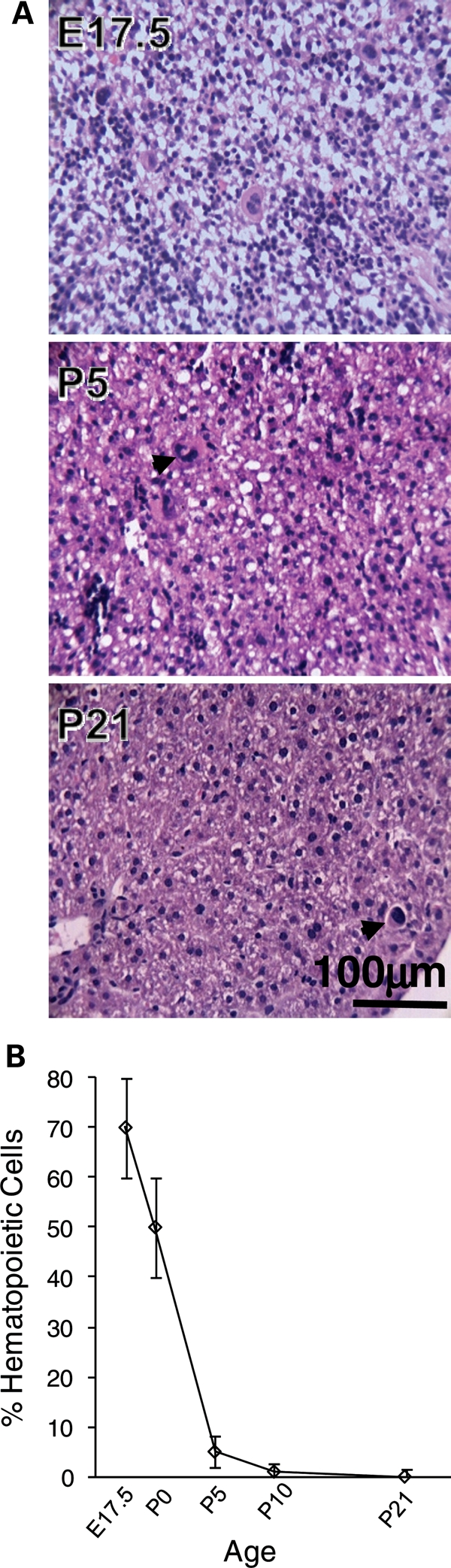
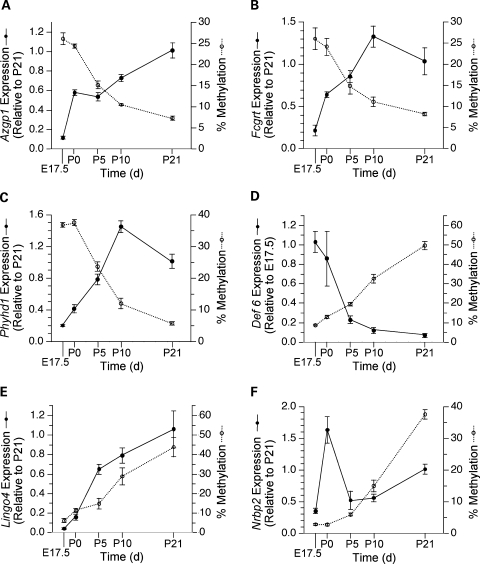
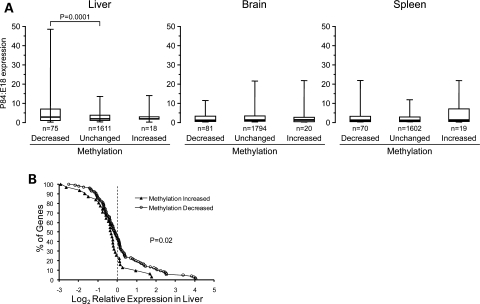
The question of whether DNA methylation contributes to the stabilization of gene expression patterns in differentiated mammalian tissues remains controversial. Using genome-wide methylation profiling, we screened 3757 gene promoters for changes in methylation during postnatal liver development to test the hypothesis that developmental changes in methylation and expression are temporally correlated. We identified 31 genes that gained methylation and 111 that lost methylation from embryonic day 17.5 to postnatal day 21. Promoters undergoing methylation changes in postnatal liver tended not to be associated with CpG islands. At most genes studied, developmental changes in promoter methylation were associated with expression changes, suggesting both that transcriptional inactivity attracts de novo methylation, and that transcriptional activity can override DNA methylation and successively induce developmental hypomethylation. These in vivo data clearly indicate a role for DNA methylation in mammalian differentiation, and provide the novel insight that critical windows in mammalian developmental epigenetics extend well beyond early embryonic development.
Figures

References
-
- Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl.):245–254. - PubMed
-
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. - PubMed
-
- Holliday R., Pugh J.E. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. - PubMed
-
- Riggs A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell. Genet. 1975;14:9–25. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
