

Acyl-CoA synthetase as a cancer survival factor: its inhibition enhances the efficacy of etoposide
- PMID: 19459852
- PMCID: PMC11158289
- DOI: 10.1111/j.1349-7006.2009.01203.x
Acyl-CoA synthetase as a cancer survival factor: its inhibition enhances the efficacy of etoposide
Abstract
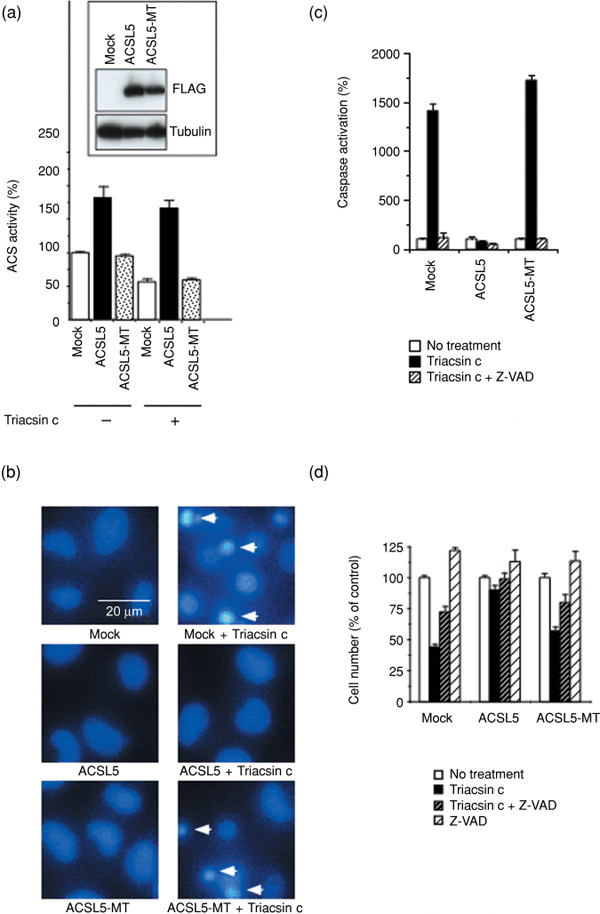
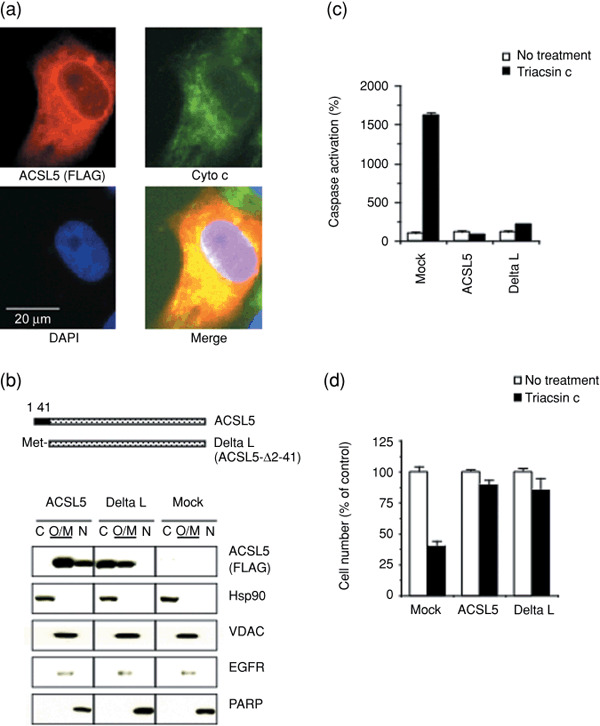
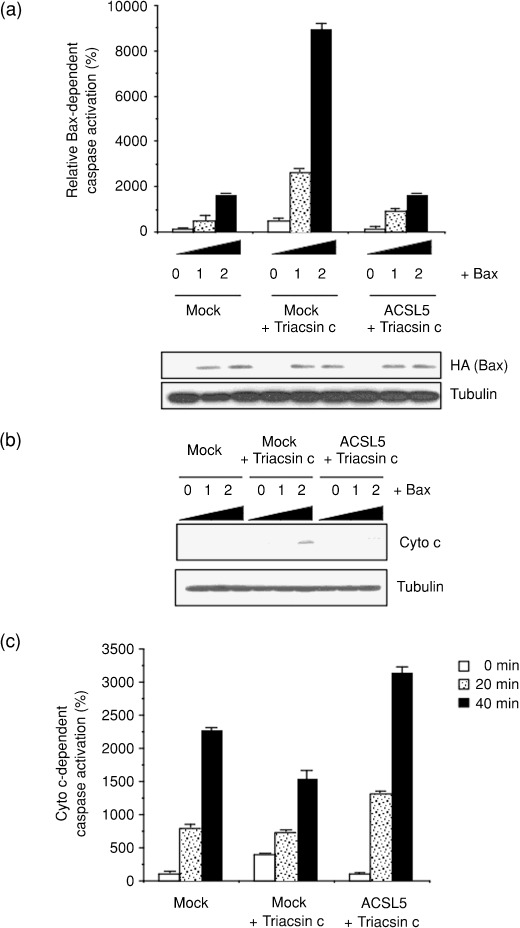
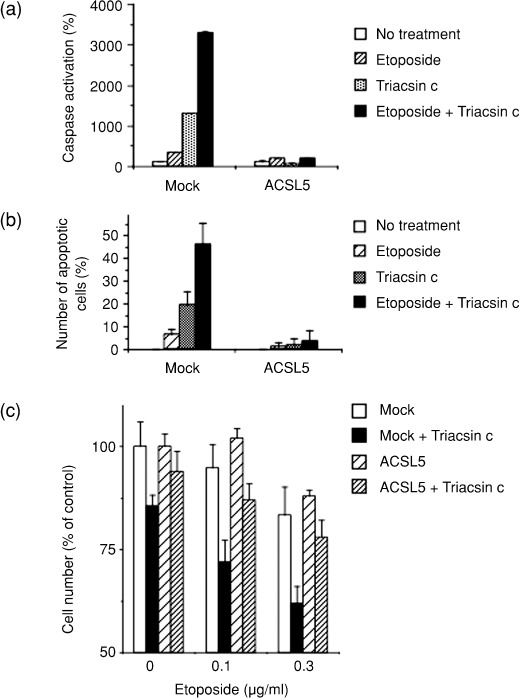
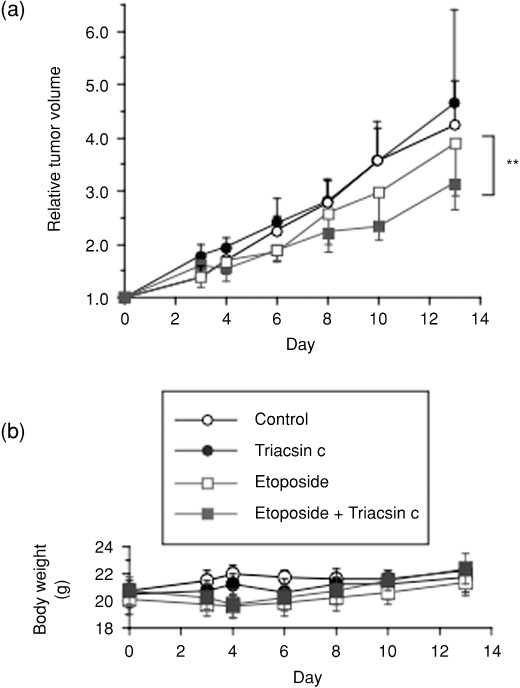
Lipid metabolism is often elevated in cancer cells and plays an important role in their growth and malignancy. Acyl-CoA synthetase (ACS), which converts long-chain fatty acids to acyl-CoA, is overexpressed in various types of cancer. However, the role of ACS in cancer remains unknown. Here, we found that ACS enzyme activity is required for cancer cell survival. Namely, the ACS inhibitor Triacsin c induced massive apoptosis in glioma cells while this cell death was completely suppressed by overexpression of ACSL5, the Triacsin c-resistant ACS isozyme, but not by overexpression of a catalytically inactive ACSL5 mutant. ACS inhibition by Triacsin c markedly potentiated the Bax-induced intrinsic apoptotic pathway by promoting cytochrome c release and subsequent caspase activation. These effects were abrogated by ACSL5 overexpression. Correspondingly, ACS inhibition synergistically potentiated the glioma cell death induced by etoposide, a well-known activator of apoptosis. Furthermore, in a nude mouse xenograft model, Triacsin c at a non-toxic dose enhanced the antitumor efficacy of a low-dose chemotherapy with etoposide. These results indicate that ACS is an apoptosis suppressor and that ACS inhibition could be a rational strategy to amplify the antitumor effect of etoposide.
Figures
Similar articles
-
p53-defective tumors with a functional apoptosome-mediated pathway: a new therapeutic target.J Natl Cancer Inst. 2005 May 18;97(10):765-77. doi: 10.1093/jnci/dji133. J Natl Cancer Inst. 2005. PMID: 15900046
-
Human intestinal acyl-CoA synthetase 5 is sensitive to the inhibitor triacsin C.World J Gastroenterol. 2011 Nov 28;17(44):4883-9. doi: 10.3748/wjg.v17.i44.4883. World J Gastroenterol. 2011. PMID: 22171129 Free PMC article.
-
Evidence for an essential role of long chain acyl-CoA synthetase in animal cell proliferation. Inhibition of long chain acyl-CoA synthetase by triacsins caused inhibition of Raji cell proliferation.J Biol Chem. 1991 Mar 5;266(7):4214-9. J Biol Chem. 1991. PMID: 1999415
-
Triacsin C: a differential inhibitor of arachidonoyl-CoA synthetase and nonspecific long chain acyl-CoA synthetase.Prostaglandins. 1989 Jun;37(6):655-71. doi: 10.1016/0090-6980(89)90103-2. Prostaglandins. 1989. PMID: 2505330
-
Lipid metabolism inhibitors of microbial origin.Kitasato Arch Exp Med. 1993 Apr;65 Suppl:1-12. Kitasato Arch Exp Med. 1993. PMID: 7967370 Review.
Cited by
-
Splice-site variant in ACSL5: a marker promoting opposing effect on cell viability and protein expression.Eur J Hum Genet. 2019 Dec;27(12):1836-1844. doi: 10.1038/s41431-019-0414-5. Epub 2019 May 3. Eur J Hum Genet. 2019. PMID: 31053784 Free PMC article.
-
Fatty acid metabolism in breast cancer subtypes.Oncotarget. 2017 Apr 25;8(17):29487-29500. doi: 10.18632/oncotarget.15494. Oncotarget. 2017. PMID: 28412757 Free PMC article. Review.
-
ACSL5 regulated acetyl-CoA to promote bladder cancer cellular senescence via 53BP1 acetylation.Oncogene. 2025 Sep;44(34):3096-3112. doi: 10.1038/s41388-025-03474-1. Epub 2025 Jul 1. Oncogene. 2025. PMID: 40595416
-
TIM-3/Gal-9 interaction affects glucose and lipid metabolism in acute myeloid leukemia cell lines.Front Immunol. 2023 Nov 10;14:1267578. doi: 10.3389/fimmu.2023.1267578. eCollection 2023. Front Immunol. 2023. PMID: 38022614 Free PMC article.
-
Developing targeted therapies for neuroblastoma by dissecting the effects of metabolic reprogramming on tumor microenvironments and progression.Theranostics. 2024 May 27;14(9):3439-3469. doi: 10.7150/thno.93962. eCollection 2024. Theranostics. 2024. PMID: 38948053 Free PMC article.
References
-
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7: 763–77. - PubMed
-
- Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res 2006; 66: 5977–80. - PubMed
-
- Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference‐mediated silencing of the acetyl‐CoA‐carboxylase‐alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res 2005; 65: 6719–25. - PubMed
-
- Coleman RA, Lewin TM, Van Horn CG, Gonzalez‐Baro MR. Do long‐chain acyl‐CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr 2002; 132: 2123–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
