

Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease
- PMID: 19460319
- PMCID: PMC3609037
- DOI: 10.1016/j.meegid.2009.02.003
Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease
Abstract
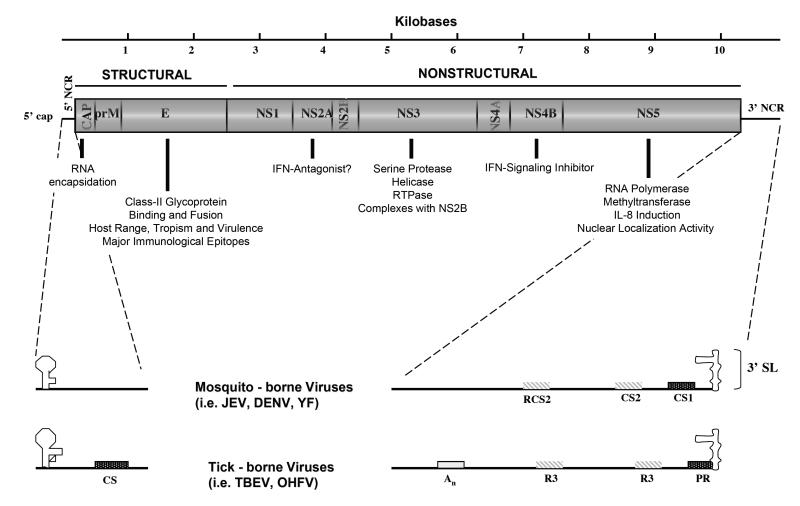
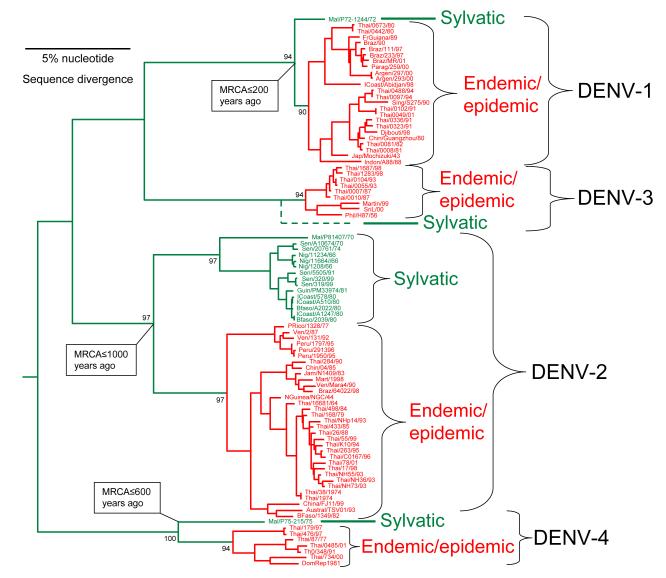
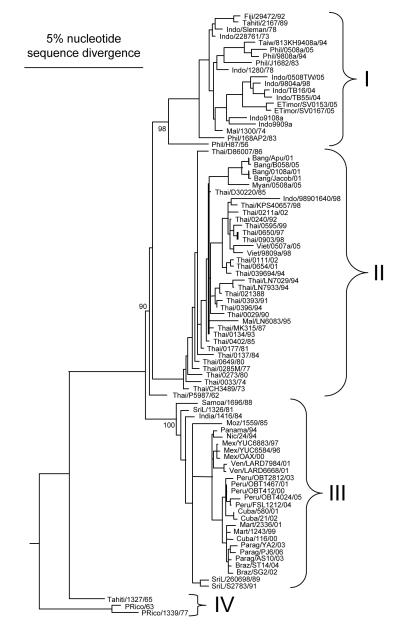
Dengue viruses (DENV) are the most important arboviral pathogens in tropical and subtropical regions throughout the world, putting at risk of infection nearly a third of the global human population. Evidence from the historical record suggests a long association between these viruses and humans. The transmission of DENV includes a sylvatic, enzootic cycle between nonhuman primates and arboreal mosquitoes of the genus Aedes, and an urban, endemic/epidemic cycle between Aedes aegypti, a mosquito with larval development in peridomestic water containers, and human reservoir hosts. DENV are members of the genus Flavivirus in the Family Flaviviridae and comprise of 4 antigenically distinct serotypes (DENV-1-4). Although they are nearly identical epidemiologically, the 4 DENV serotypes are genetically quite distinct. Utilization of phylogenetic analyses based on partial and/or complete genomic sequences has elucidated the origins, epidemiology (genetic diversity, transmission dynamics and epidemic potential), and the forces that shape DENV molecular evolution (rates of evolution, selection pressures, population sizes, putative recombination and evolutionary constraints) in nature. In this review, we examine how phylogenetics have improved understanding of DENV population dynamics and sizes at various stages of infection and transmission, and how this information may influence pathogenesis and improve our ability to understand and predict DENV emergence.
Figures




References
-
- Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science. 2006;311(5758):236–8. - PubMed
-
- AbuBakar S, Wong PF, Chan YF. Emergence of dengue virus type 4 genotype IIA in Malaysia. J Gen Virol. 2002;83(Pt 10):2437–42. - PubMed
-
- Ackermann M, Padmanabhan R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J Biol Chem. 2001;276(43):39926–37. - PubMed
-
- Ali M, Wagatsuma Y, Emch M, Breiman RF. Use of a geographic information system for defining spatial risk for dengue transmission in Bangladesh: role for Aedes albopictus in an urban outbreak. Am. J. Trop. Med. Hyg. 2003;69(6):634–40. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
