

Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase
- PMID: 19461581
- PMCID: PMC2699363
- DOI: 10.1038/emboj.2009.138
Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase
Abstract
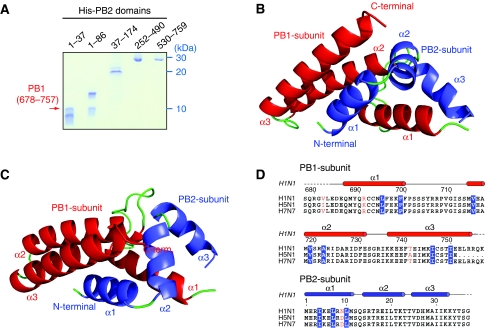
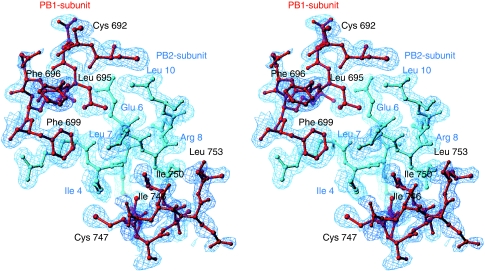
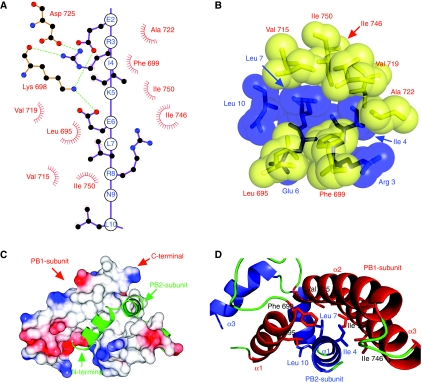
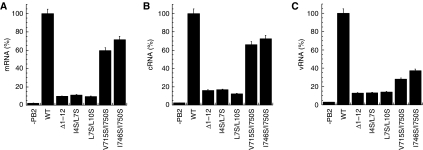
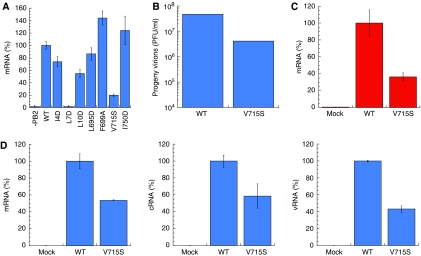
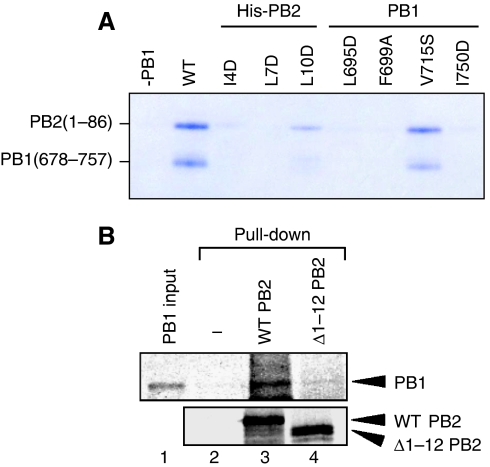
Influenza virus RNA-dependent RNA polymerase is a multi-functional heterotrimer, which uses a 'cap-snatching' mechanism to produce viral mRNA. Host cell mRNA is cleaved to yield a cap-bearing oligonucleotide, which can be extended using viral genomic RNA as a template. The cap-binding and endonuclease activities are only activated once viral genomic RNA is bound. This requires signalling from the RNA-binding PB1 subunit to the cap-binding PB2 subunit, and the interface between these two subunits is essential for the polymerase activity. We have defined this interaction surface by protein crystallography and tested the effects of mutating contact residues on the function of the holo-enzyme. This novel interface is surprisingly small, yet, it has a crucial function in regulating the 250 kDa polymerase complex and is completely conserved among avian and human influenza viruses.
Figures
References
-
- Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW (2009) The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458: 914–918 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
