

Selective plane illumination microscopy techniques in developmental biology
- PMID: 19465594
- PMCID: PMC2685720
- DOI: 10.1242/dev.022426
Selective plane illumination microscopy techniques in developmental biology
Abstract
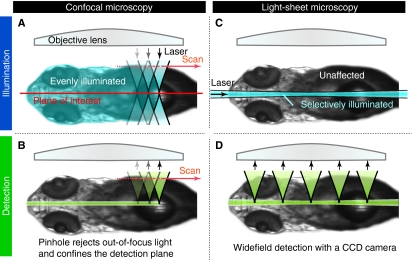
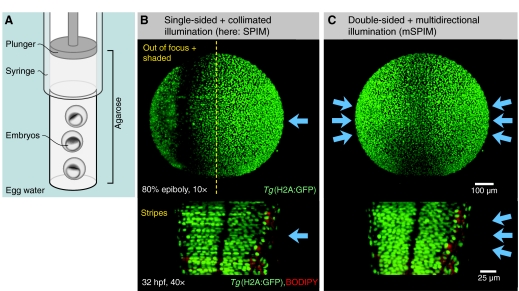
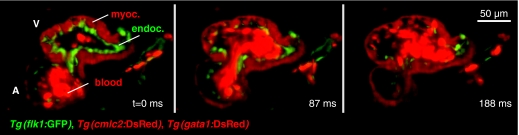
Selective plane illumination microscopy (SPIM) and other fluorescence microscopy techniques in which a focused sheet of light serves to illuminate the sample have become increasingly popular in developmental studies. Fluorescence light-sheet microscopy bridges the gap in image quality between fluorescence stereomicroscopy and high-resolution imaging of fixed tissue sections. In addition, high depth penetration, low bleaching and high acquisition speeds make light-sheet microscopy ideally suited for extended time-lapse experiments in live embryos. This review compares the benefits and challenges of light-sheet microscopy with established fluorescence microscopy techniques such as confocal microscopy and discusses the different implementations and applications of this easily adaptable technology.
Figures





References
-
- Becker, K., Jährling, N., Kramer, E. R., Schnorrer, F. and Dodt, H.-U. (2008). Ultramicroscopy: 3D reconstruction of large microscopical specimens. J. Biophotonics 1, 36-42. - PubMed
-
- Breuninger, T., Greger, K. and Stelzer, E. H. K. (2007). Lateral modulation boosts image quality in single plane illumination fluorescence microscopy. Opt. Lett. 32, 1938-1940. - PubMed
-
- Buytaert, J. A. N. and Dirckx, J. J. J. (2007). Design and quantitative resolution measurements of an optical virtual sectioning three-dimensional imaging technique for biomedical specimens, featuring two-micrometer slicing resolution. J. Biomed. Opt. 12, 014039. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
