

Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria
- PMID: 19465910
- PMCID: PMC2883584
- DOI: 10.1038/ng.380
Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria
Abstract
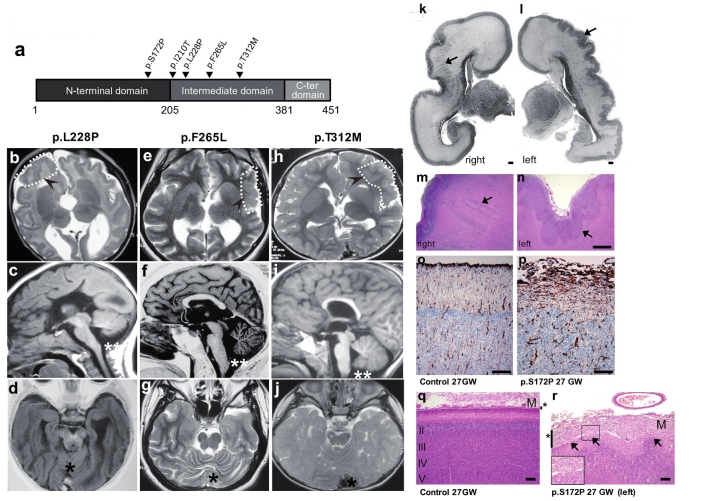
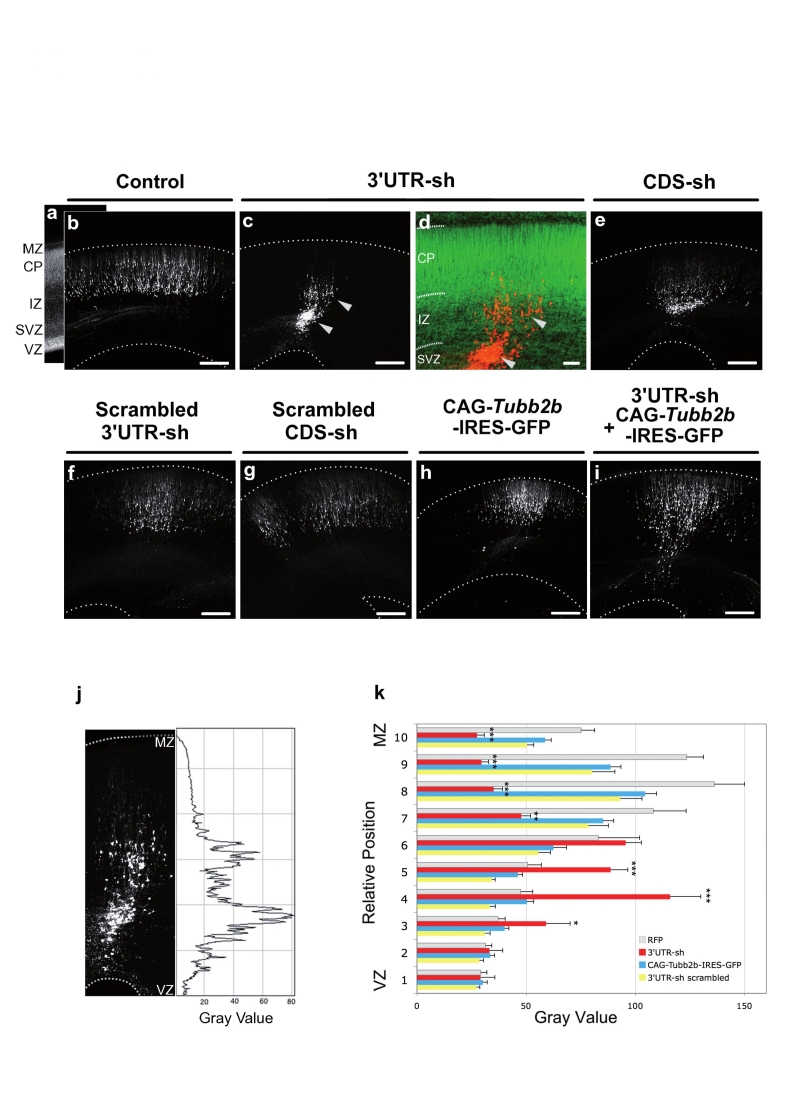
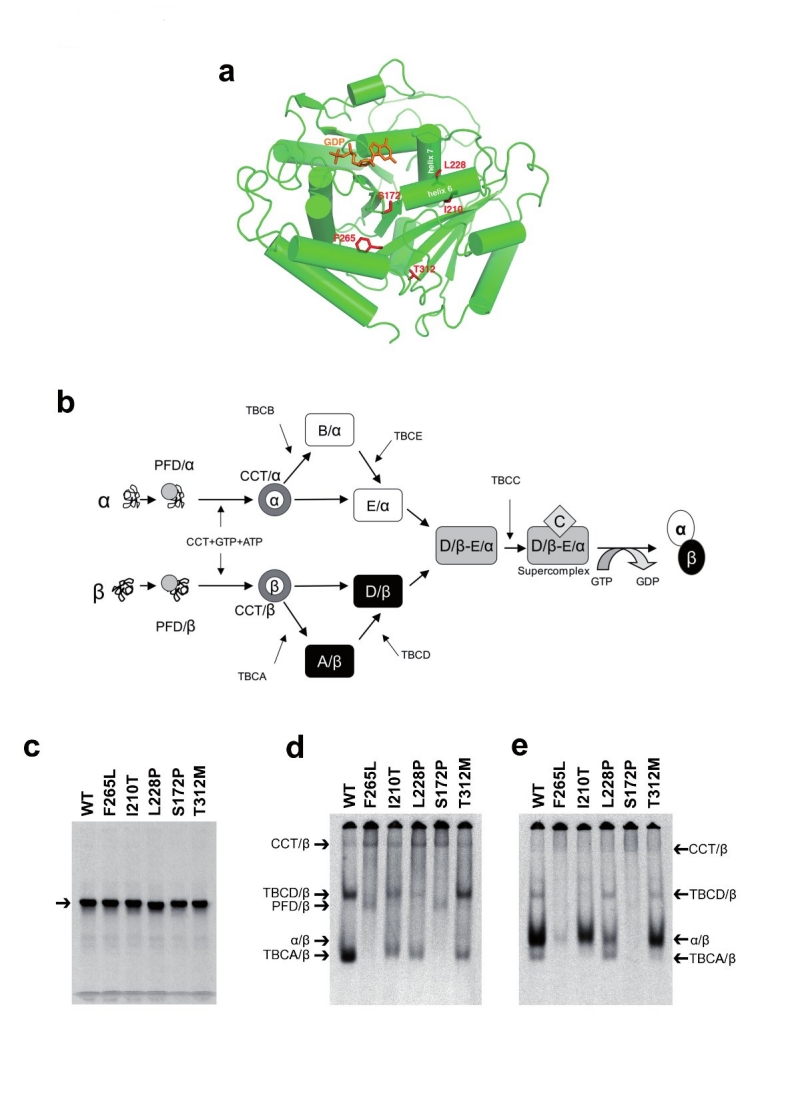
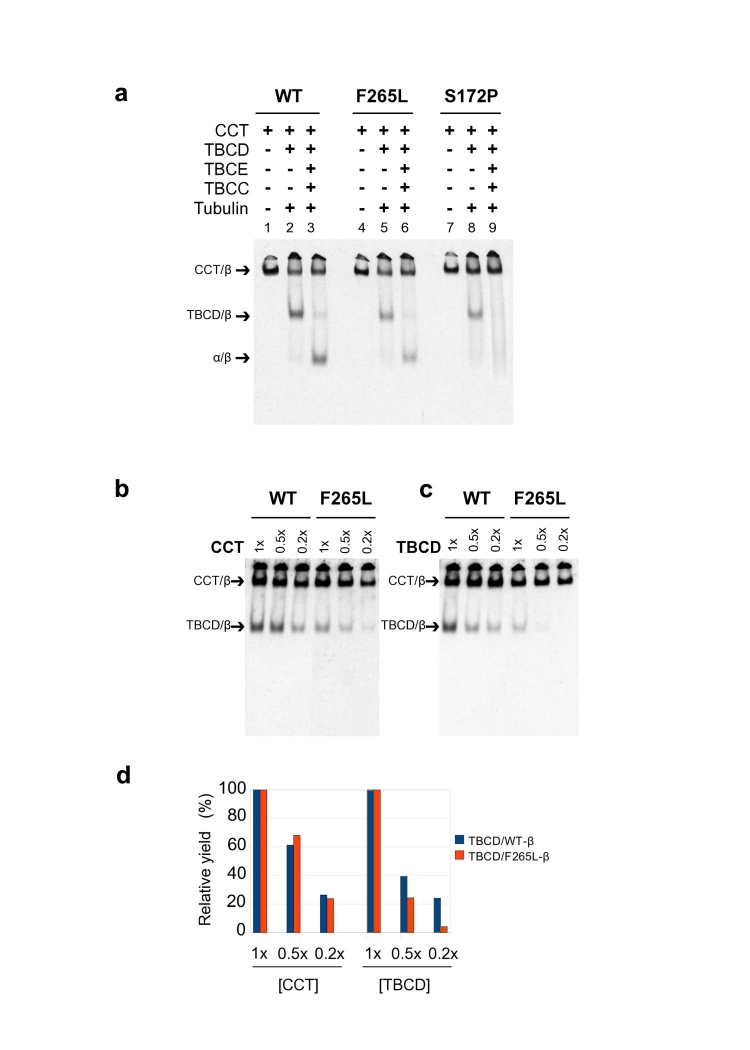
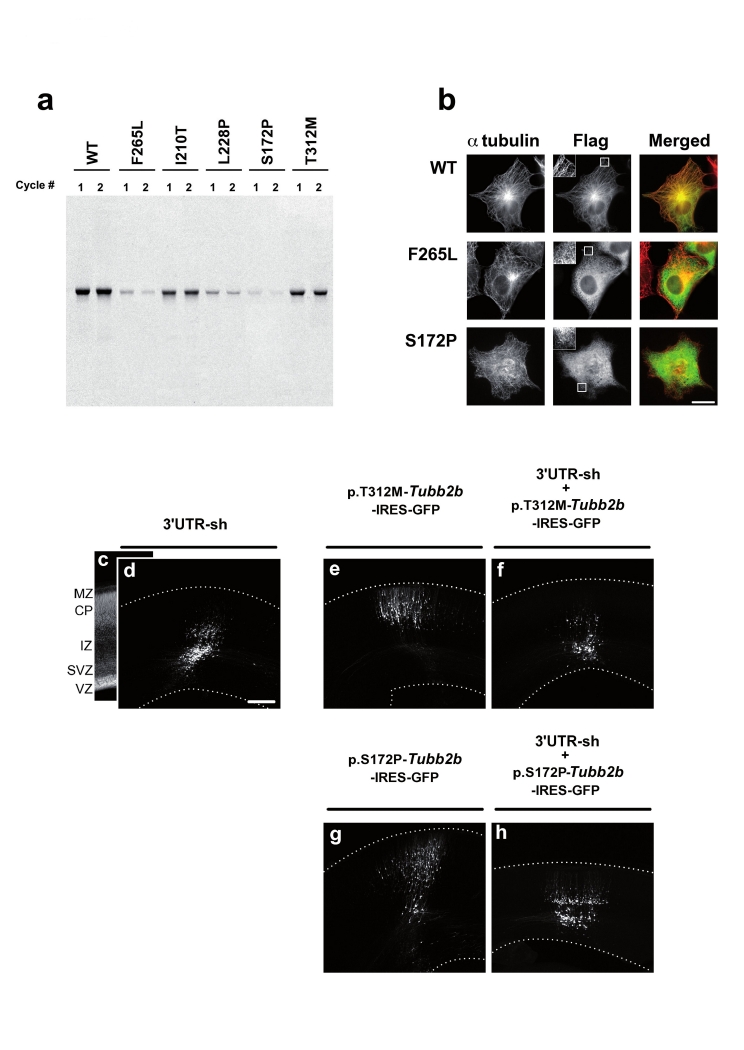
Polymicrogyria is a relatively common but poorly understood defect of cortical development characterized by numerous small gyri and a thick disorganized cortical plate lacking normal lamination. Here we report de novo mutations in a beta-tubulin gene, TUBB2B, in four individuals and a 27-gestational-week fetus with bilateral asymmetrical polymicrogyria. Neuropathological examination of the fetus revealed an absence of cortical lamination associated with the presence of ectopic neuronal cells in the white matter and in the leptomeningeal spaces due to breaches in the pial basement membrane. In utero RNAi-based inactivation demonstrates that TUBB2B is required for neuronal migration. We also show that two disease-associated mutations lead to impaired formation of tubulin heterodimers. These observations, together with previous data, show that disruption of microtubule-based processes underlies a large spectrum of neuronal migration disorders that includes not only lissencephaly and pachygyria, but also polymicrogyria malformations.
Figures
Comment in
-
Diversifying microtubules in brain development.Nat Genet. 2009 Jun;41(6):638-40. doi: 10.1038/ng0609-638. Nat Genet. 2009. PMID: 19471300 No abstract available.
References
-
- Dutcher SK. The tubulin fraternity: alpha to eta. Curr Opin Cell Biol. 2001;13:49–54. - PubMed
-
- Poirier K, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A(TUBA1A). Hum Mutat. 2007;28:1055–64. - PubMed
-
- Bahi-Buisson N, et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J Med Genet. 2008;45:647–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
