

Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models
- PMID: 19468344
- PMCID: PMC2680652
- DOI: 10.3390/ijms10041872
Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models
Abstract
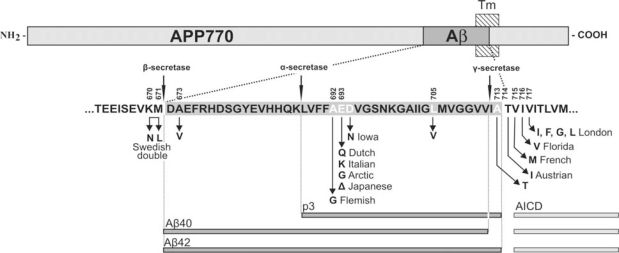
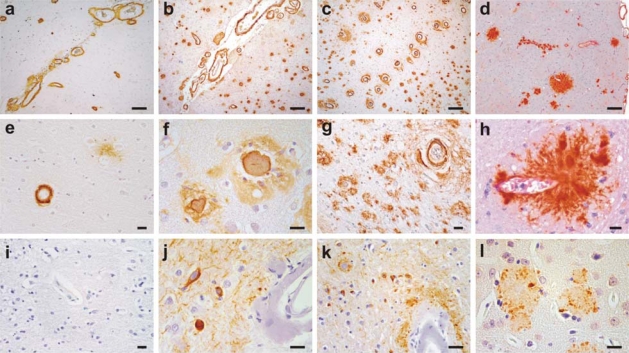
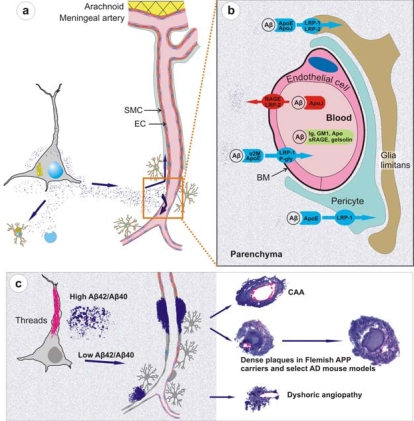
Cerebral amyloid angiopathy (CAA) refers to the specific deposition of amyloid fibrils in the leptomeningeal and cerebral blood vessel walls, often causing secondary vascular degenerative changes. Although many kinds of peptides are known to be deposited as vascular amyloid, amyloid-beta (Abeta)-CAA is the most common type associated with normal aging, sporadic CAA, Alzheimer's disease (AD) and Down's syndrome. Moreover, Abeta-CAA is also associated with rare hereditary cerebrovascular amyloidosis due to mutations within the Abeta domain of the amyloid precursor protein (APP) such as Dutch and Flemish APP mutations. Genetics and clinicopathological studies on these familial diseases as well as sporadic conditions have already shown that CAA not only causes haemorrhagic and ischemic strokes, but also leads to progressive dementia. Transgenic mouse models based on familial AD mutations have also successfully reproduced many of the features found in human disease, providing us with important insights into the pathogenesis of CAA. Importantly, such studies have pointed out that specific vastopic Abeta variants or an unaltered Abeta42/Abeta40 ratio favor vascular Abeta deposition over parenchymal plaques, but higher than critical levels of Abeta40 are also observed to be anti-amyloidogenic. These data would be important in the development of therapies targeting amyloid in vessels.
Keywords: Alzheimer’s disease; CAA; amyloid β protein; cerebrovascular amyloidosis; dense-core plaques; pathogenesis; senile plaques; therapy; transgenic mice.
Figures
Similar articles
-
Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques.Genes Brain Behav. 2008 Feb;7 Suppl 1:67-82. doi: 10.1111/j.1601-183X.2007.00380.x. Genes Brain Behav. 2008. PMID: 18184371 Review.
-
Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons.Am J Pathol. 2000 Oct;157(4):1283-98. doi: 10.1016/S0002-9440(10)64644-5. Am J Pathol. 2000. PMID: 11021833 Free PMC article.
-
Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models.Acta Neuropathol Commun. 2015 Nov 10;3:70. doi: 10.1186/s40478-015-0250-y. Acta Neuropathol Commun. 2015. PMID: 26556230 Free PMC article.
-
Amyloid-β peptide signature associated with cerebral amyloid angiopathy in familial Alzheimer's disease with APPdup and Down syndrome.Acta Neuropathol. 2024 Jul 18;148(1):8. doi: 10.1007/s00401-024-02756-4. Acta Neuropathol. 2024. PMID: 39026031 Free PMC article.
-
Cerebral amyloid angiopathy and dementia.Panminerva Med. 2004 Dec;46(4):253-64. Panminerva Med. 2004. PMID: 15876981 Review.
Cited by
-
Emerging concepts in sporadic cerebral amyloid angiopathy.Brain. 2017 Jul 1;140(7):1829-1850. doi: 10.1093/brain/awx047. Brain. 2017. PMID: 28334869 Free PMC article. Review.
-
Murine models of Alzheimer's disease and their use in developing immunotherapies.Biochim Biophys Acta. 2010 Oct;1802(10):847-59. doi: 10.1016/j.bbadis.2010.05.004. Epub 2010 May 13. Biochim Biophys Acta. 2010. PMID: 20471477 Free PMC article. Review.
-
Blocking the apolipoprotein E/amyloid-β interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice.J Alzheimers Dis. 2011;24(2):269-85. doi: 10.3233/JAD-2011-101401. J Alzheimers Dis. 2011. PMID: 21239853 Free PMC article.
-
Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ APP mutation.Transl Psychiatry. 2012 Nov 13;2(11):e183. doi: 10.1038/tp.2012.109. Transl Psychiatry. 2012. PMID: 23149447 Free PMC article.
-
Matrix metalloproteinase 2 (MMP-2) degrades soluble vasculotropic amyloid-beta E22Q and L34V mutants, delaying their toxicity for human brain microvascular endothelial cells.J Biol Chem. 2010 Aug 27;285(35):27144-27158. doi: 10.1074/jbc.M110.135228. Epub 2010 Jun 24. J Biol Chem. 2010. PMID: 20576603 Free PMC article.
References
-
- Vinters HV, Wang ZZ, Secor DL. Brain parenchymal and microvascular amyloid in Alzheimer's disease. Brain Pathol. 1996;6:179–195. - PubMed
-
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J. Neural Transm. 2002;109:813–836. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Scholtz W. Studien zur Pathologie der Hirngefässe. II. Die drüsige Entartung der Hirnarterien und -capillären. Z.Gesamte.Neurol.Psychiat. 1938;162:694–715.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
