

An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy
- PMID: 19470612
- PMCID: PMC2707104
- DOI: 10.1242/dmm.002527
An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy
Abstract
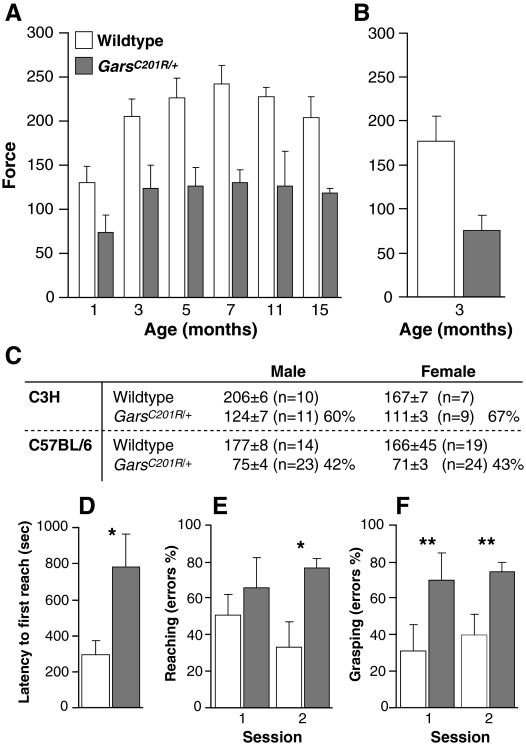
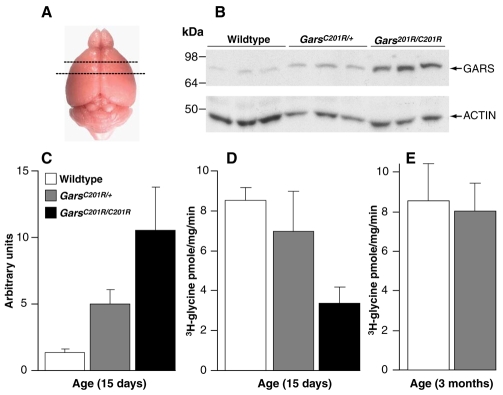
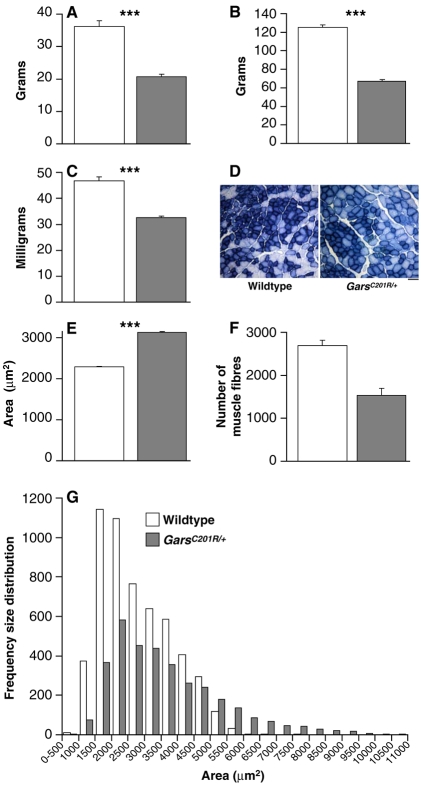
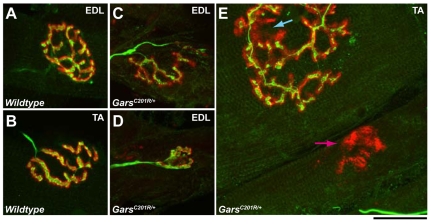
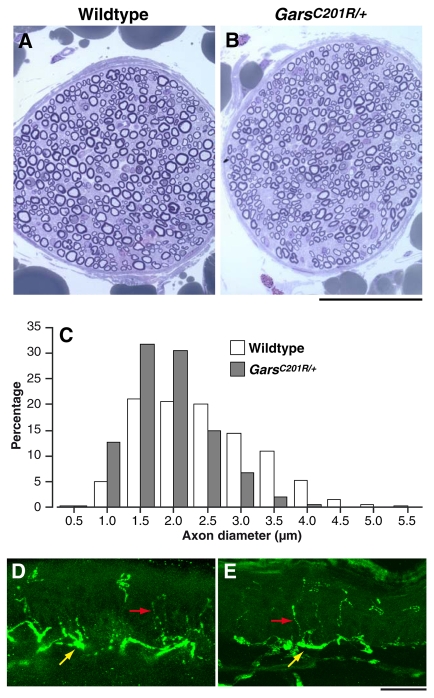
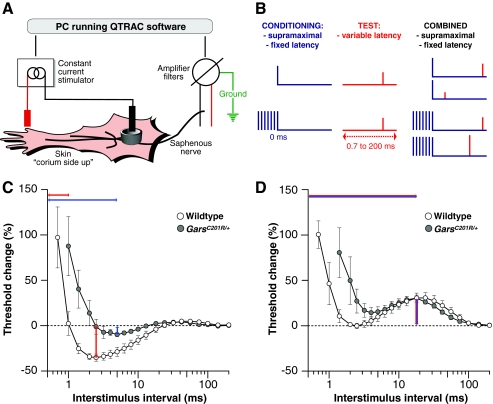
Mutations in the enzyme glycyl-tRNA synthetase (GARS) cause motor and sensory axon loss in the peripheral nervous system in humans, described clinically as Charcot-Marie-Tooth type 2D or distal spinal muscular atrophy type V. Here, we characterise a new mouse mutant, Gars(C201R), with a point mutation that leads to a non-conservative substitution within GARS. Heterozygous mice with a C3H genetic background have loss of grip strength, decreased motor flexibility and disruption of fine motor control; this relatively mild phenotype is more severe on a C57BL/6 background. Homozygous mutants have a highly deleterious set of features, including movement difficulties and death before weaning. Heterozygous animals have a reduction in axon diameter in peripheral nerves, slowing of nerve conduction and an alteration in the recovery cycle of myelinated axons, as well as innervation defects. An assessment of GARS levels showed increased protein in 15-day-old mice compared with controls; however, this increase was not observed in 3-month-old animals, indicating that GARS function may be more crucial in younger animals. We found that enzyme activity was not reduced detectably in heterozygotes at any age, but was diminished greatly in homozygous mice compared with controls; thus, homozygous animals may suffer from a partial loss of function. The Gars(C201R) mutation described here is a contribution to our understanding of the mechanism by which mutations in tRNA synthetases, which are fundamentally important, ubiquitously expressed enzymes, cause axonopathy in specific sets of neurons.
Figures

References
-
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, et al. (2003). Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 72, 1293–1299 - PMC - PubMed
-
- Bostock H. (2004). Nerve excitability studies: past, present, future? Suppl Clin Neurophysiol. 57, 85–90 - PubMed
-
- Cader MZ, Ren J, James PA, Bird LE, Talbot K, Stammers DK. (2007). Crystal structure of human wildtype and S581L-mutant glycyl-tRNA synthetase: an enzyme underlying distal spinal muscular atrophy. FEBS Lett. 581, 2959–2964 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
