

Alteration of enzyme specificity by computational loop remodeling and design
- PMID: 19470646
- PMCID: PMC2685249
- DOI: 10.1073/pnas.0811070106
Alteration of enzyme specificity by computational loop remodeling and design
Abstract
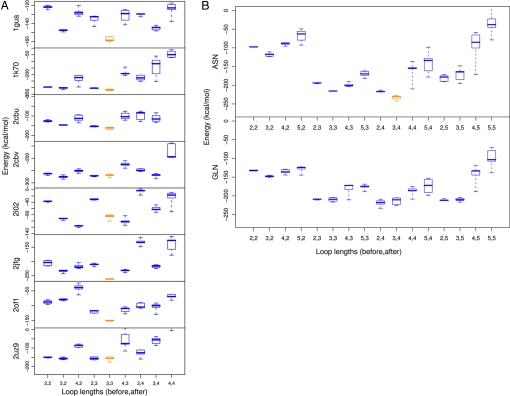
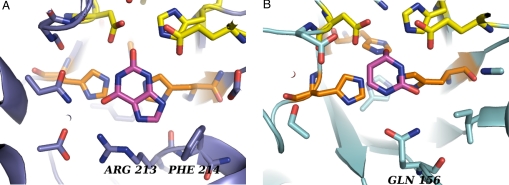
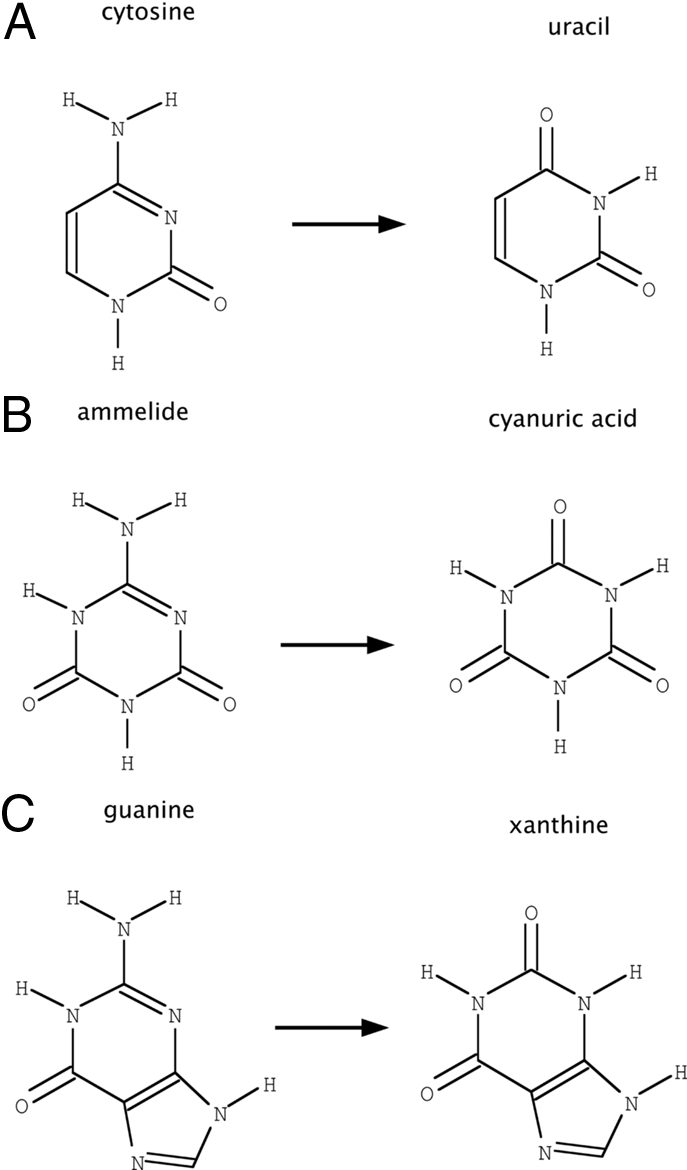
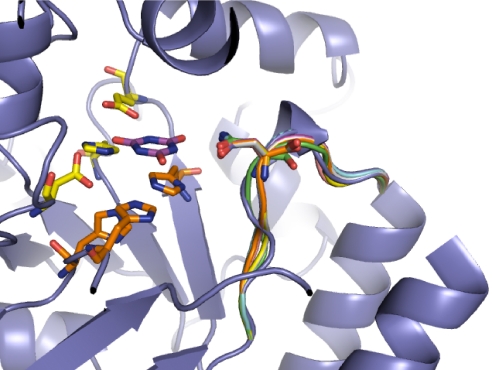
Altering the specificity of an enzyme requires precise positioning of side-chain functional groups that interact with the modified groups of the new substrate. This requires not only sequence changes that introduce the new functional groups but also sequence changes that remodel the structure of the protein backbone so that the functional groups are properly positioned. We describe a computational design method for introducing specific enzyme-substrate interactions by directed remodeling of loops near the active site. Benchmark tests on 8 native protein-ligand complexes show that the method can recover native loop lengths and, often, native loop conformations. We then use the method to redesign a critical loop in human guanine deaminase such that a key side-chain interaction is made with the substrate ammelide. The redesigned enzyme is 100-fold more active on ammelide and 2.5e4-fold less active on guanine than wild-type enzyme: The net change in specificity is 2.5e6-fold. The structure of the designed protein was confirmed by X-ray crystallographic analysis: The remodeled loop adopts a conformation that is within 1-A Calpha RMSD of the computational model.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Dahiyat BI, Mayo SL. De novo protein design: Fully automated sequence selection. Science. 1997;278:82–87. - PubMed
-
- Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. - PubMed
-
- Looger LL, et al. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423:185–190. - PubMed
-
- Kortemme T, et al. Computational redesign of protein–protein interaction specificity. Nat Struct Mol Biol. 2004;11:371–379. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
