

HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer
- PMID: 19470725
- PMCID: PMC3159511
- DOI: 10.1158/1078-0432.CCR-08-3252
HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer
Abstract
Purpose: We determined hepatocyte growth factor (HGF) and c-Met expression and signaling in human head and neck squamous cell carcinoma (HNSCC) cells and primary tissues and tested the ability of c-Met tyrosine kinase inhibitors (TKI) to block HGF-induced biological signaling.
Experimental design: Expression and signaling were determined using immunoblotting, ELISA, and immunohistochemistry. Biological end points included wound healing, cell proliferation, and invasion. c-Met TKIs were tested for their ability to block HGF-induced signaling and biological effects in vitro and in xenografts established in nude mice.
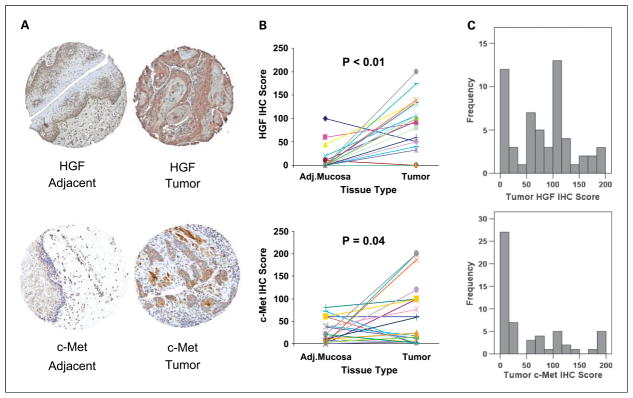
Results: c-Met was expressed and functional in HNSCC cells. HGF was secreted by HNSCC tumor-derived fibroblasts, but not by HNSCC cells. Activation of c-Met promoted phosphorylation of AKT and mitogen-activated protein kinase as well as release of the inflammatory cytokine interleukin-8. Cell growth and wound healing were also stimulated by HGF. c-Met TKIs blocked HGF-induced signaling, interleukin-8 release, and wound healing. Enhanced invasion of HNSCC cells induced by the presence of tumor-derived fibroblasts was completely blocked with a HGF-neutralizing antibody. PF-2341066, a c-Met TKI, caused a 50% inhibition of HNSCC tumor growth in vivo with decreased proliferation and increased apoptosis within the tumors. In HNSCC tumor tissues, both HGF and c-Met protein were increased compared with expression in normal mucosa.
Conclusions: These results show that HGF acts mainly as a paracrine factor in HNSCC cells, the HGF/c-Met pathway is frequently up-regulated and functional in HNSCC, and a clinically relevant c-Met TKI shows antitumor activity in vivo. Blocking the HGF/c-Met pathway may be clinically useful for the treatment of HNSCC.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures





References
-
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. - PubMed
-
- Molina JR, Adjei AA. The Ras/Raf/MAPK pathway. J Thorac Oncol. 2006;1:7–9. - PubMed
-
- Lee KH, Choi EY, Hyun MS, et al. Association of extracellular cleavage of E-cadherin mediated by MMP-7 with HGF-induced in vitro invasion in human stomach cancer cells. Eur Surg Res. 2007;39:208–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
