

Neural deficits contribute to respiratory insufficiency in Pompe disease
- PMID: 19474295
- PMCID: PMC2695054
- DOI: 10.1073/pnas.0902534106
Neural deficits contribute to respiratory insufficiency in Pompe disease
Abstract
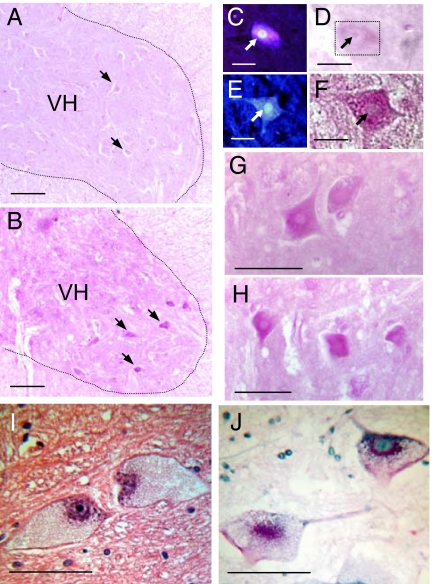
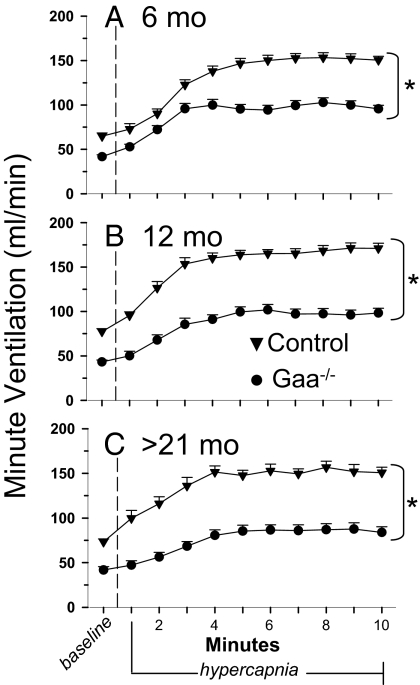
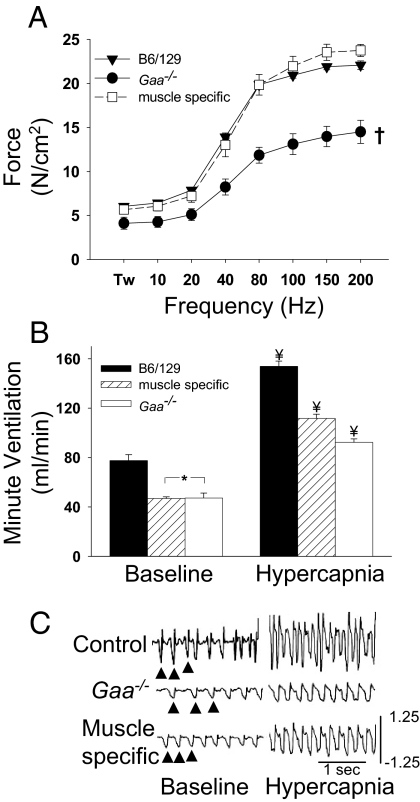
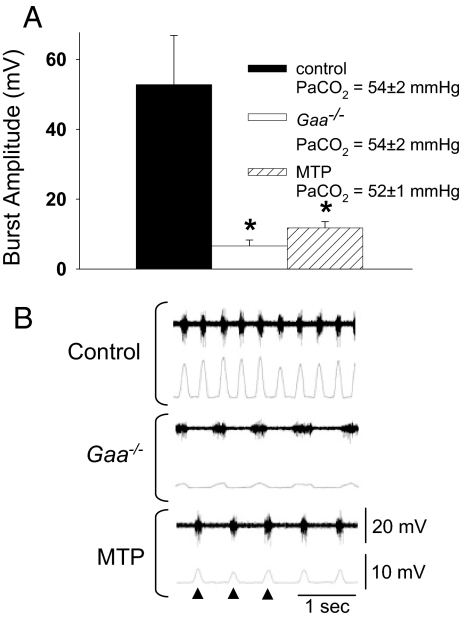
Pompe disease is a severe form of muscular dystrophy due to glycogen accumulation in all tissues, especially striated muscle. Disease severity is directly related to the deficiency of acid alpha-glucosidase (GAA), which degrades glycogen in the lysosome. Respiratory dysfunction is a hallmark of the disease, muscle weakness has been viewed as the underlying cause, and the possibility of an associated neural contribution has not been evaluated previously. Therefore, we examined behavioral and neurophysiological aspects of breathing in 2 animal models of Pompe disease--the Gaa(-/-) mouse and a transgenic line (MTP) expressing GAA only in skeletal muscle, as well as a detailed analysis of the CNS in a Pompe disease patient. Glycogen content was elevated in the Gaa(-/-) mouse cervical spinal cord. Retrograde labeling of phrenic motoneurons showed significantly greater soma size in Gaa(-/-) mice vs. isogenic controls, and glycogen was observed in Gaa(-/-) phrenic motoneurons. Ventilation, assessed via plethysmography, was attenuated during quiet breathing and hypercapnic challenge in Gaa(-/-) mice (6 to >21 months of age) vs. controls. We confirmed that MTP mice had normal diaphragmatic contractile properties; however, MTP mice had ventilation similar to the Gaa(-/-) mice during quiet breathing. Neurophysiological recordings indicated that efferent phrenic nerve inspiratory burst amplitudes were substantially lower in Gaa(-/-) and MTP mice vs. controls. In human samples, we demonstrated similar pathology in the cervical spinal cord and greater accumulation of glycogen in spinal cord compared with brain. We conclude that neural output to the diaphragm is deficient in Gaa(-/-) mice, and therapies targeting muscle alone may be ineffective in Pompe disease.
Conflict of interest statement
Conflict of interest statement: B.J.B., The Johns Hopkins University, and the University of Florida could be entitled to patent royalties for inventions described in this manuscript.
Figures
References
-
- Brown AM, Baltan Tekkök S, Ransom BR. Energy transfer from astrocytes to axons: The role of CNS glycogen. Neurochem Int. 2004;45:529–536. - PubMed
-
- Vilchez D, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–1413. - PubMed
-
- Hirschhorn R, Reuser AJJ. Glycogen storage disease type II; acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. 8th Ed. New York: McGraw-Hill; 2001. Chapter 135.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
