

Neurofibromatosis type 2
- PMID: 19476995
- PMCID: PMC4748851
- DOI: 10.1016/S0140-6736(09)60259-2
Neurofibromatosis type 2
Abstract
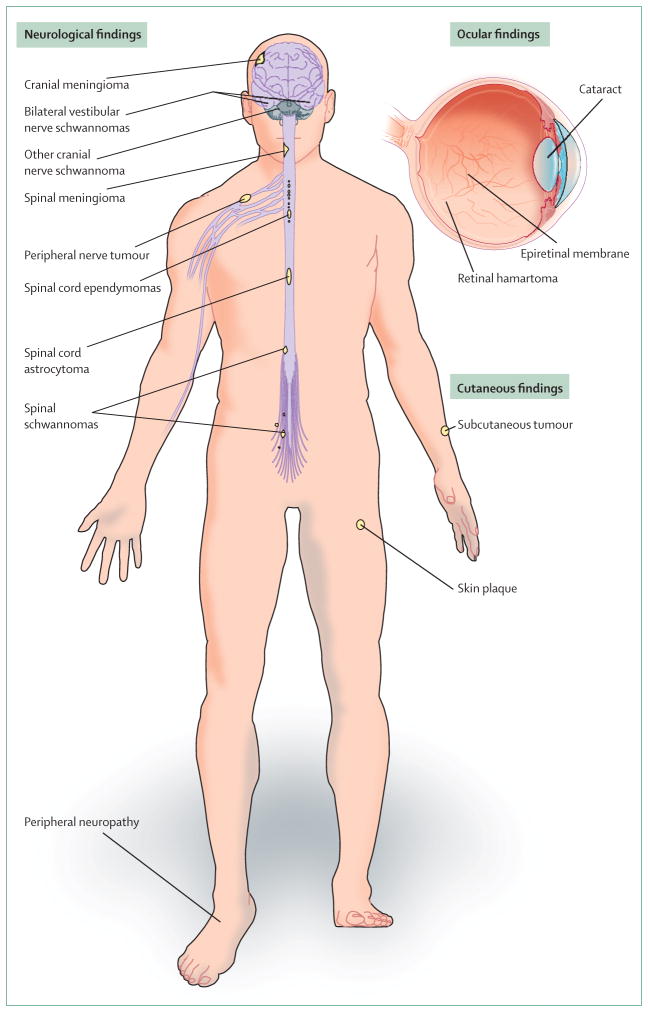
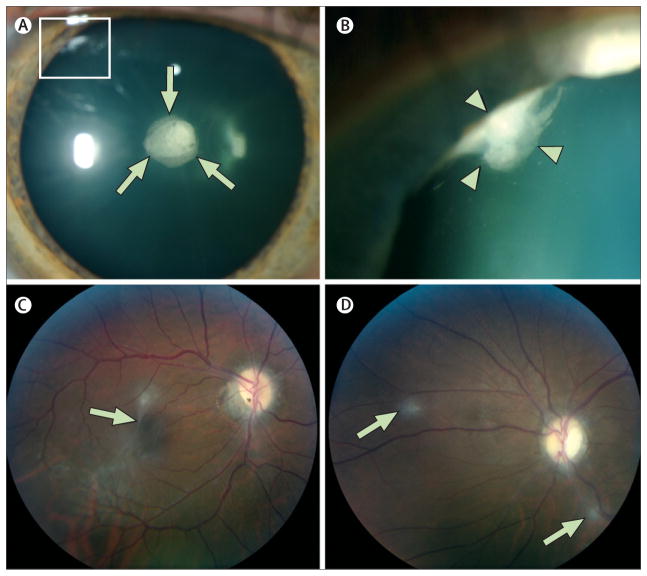
Neurofibromatosis type 2 is an autosomal-dominant multiple neoplasia syndrome that results from mutations in the NF2 tumour suppressor gene located on chromosome 22q. It has a frequency of one in 25,000 livebirths and nearly 100% penetrance by 60 years of age. Half of patients inherit a germline mutation from an affected parent and the remainder acquire a de novo mutation for neurofibromatosis type 2. Patients develop nervous system tumours (schwannomas, meningiomas, ependymomas, astrocytomas, and neurofibromas), peripheral neuropathy, ophthalmological lesions (cataracts, epiretinal membranes, and retinal hamartomas), and cutaneous lesions (skin tumours). Optimum treatment is multidisciplinary because of the complexities associated with management of the multiple, progressive, and protean lesions associated with the disorder. We review the molecular pathogenesis, genetics, clinical findings, and management strategies for neurofibromatosis type 2.
Conflict of interest statement
We declare that we have no conflicts of interest.
Figures





References
-
- Evans DGR, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26:93–97. - PubMed
-
- Evans DGR, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–18. - PubMed
-
- Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2(NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994;52:450–61. - PubMed
-
- Mautner VF, Lindenau M, Baser ME, et al. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996;38:880–86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
