

Immune pathways for translating viral infection into chronic airway disease
- PMID: 19477323
- PMCID: PMC3010226
- DOI: 10.1016/S0065-2776(09)01205-X
Immune pathways for translating viral infection into chronic airway disease
Abstract
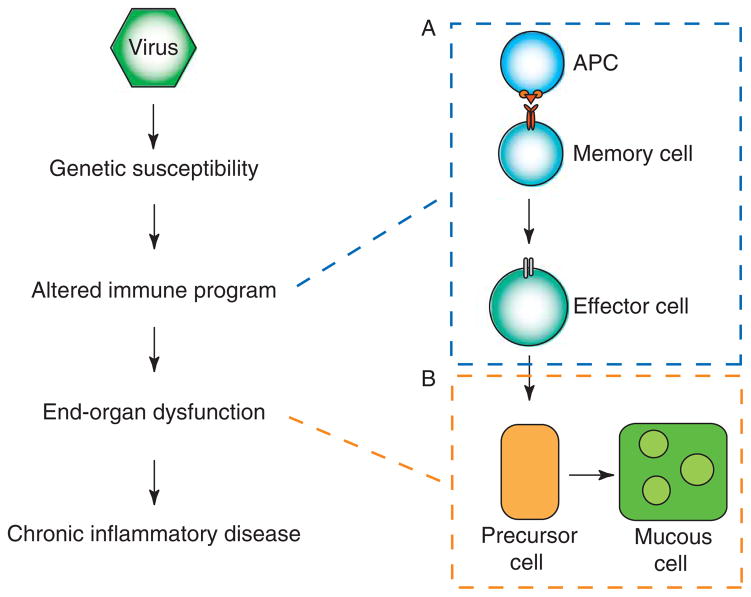
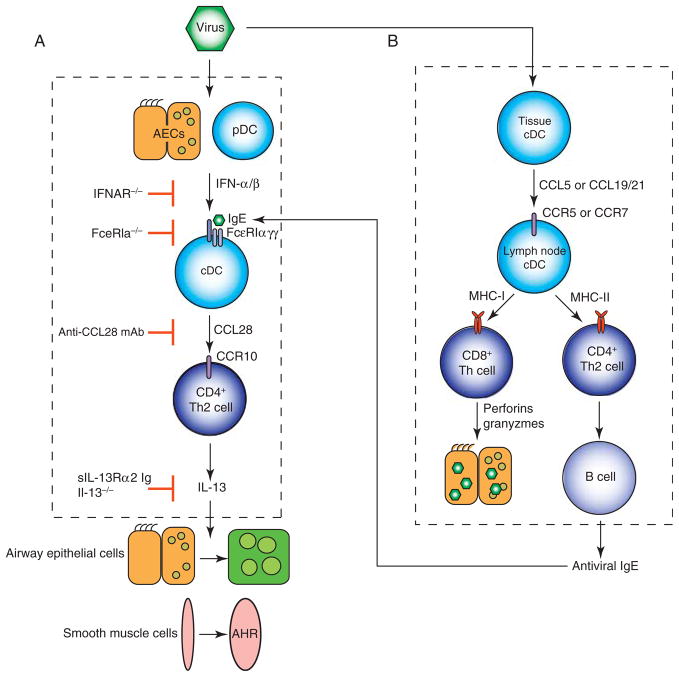
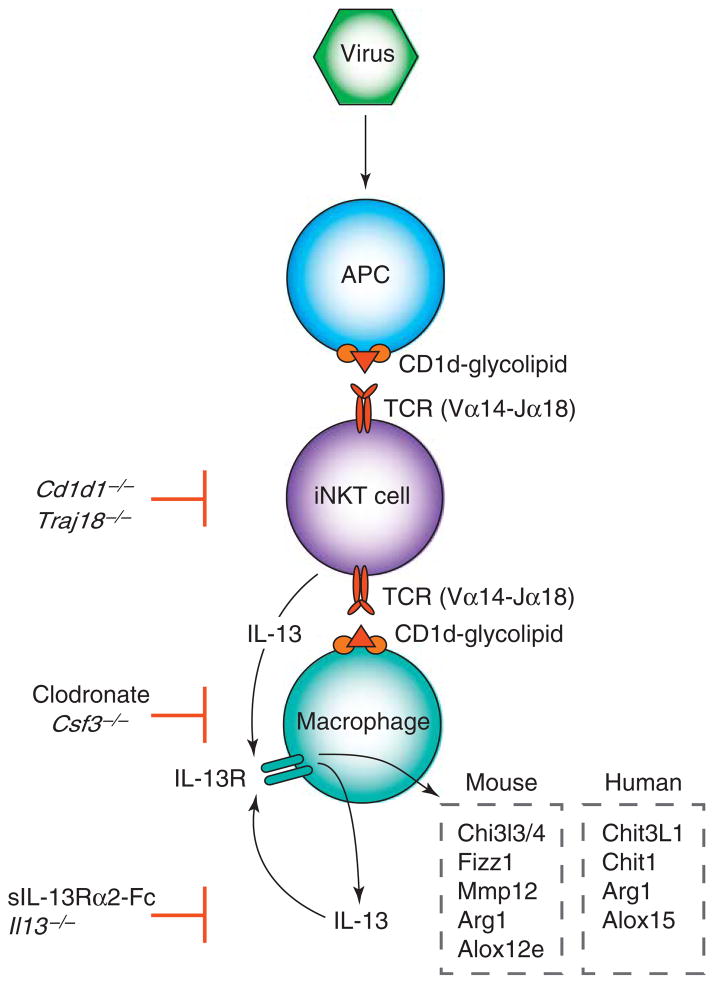
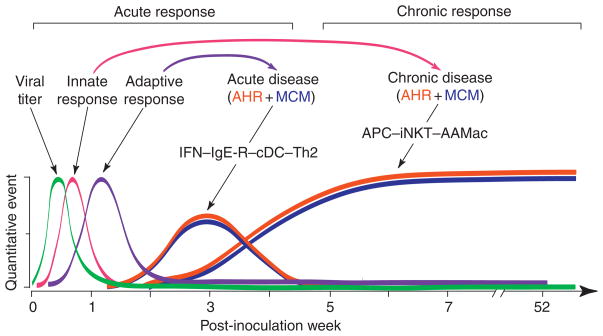
To better understand the immune basis for chronic inflammatory lung disease, we analyzed a mouse model of lung disease that develops after respiratory viral infection. The disease that develops in this model is similar to asthma and chronic obstructive pulmonary disease (COPD) in humans and is manifested after the inciting virus has been cleared to trace levels. The model thereby mimics the relationship of paramyxoviral infection to the development of childhood asthma in humans. When the acute lung disease appears in this model (at 3 weeks after viral inoculation), it depends on an immune axis that is initiated by expression and activation of the high-affinity IgE receptor (FcvarepsilonRI) on conventional lung dendritic cells (cDCs) to recruit interleukin (IL)-13-producing CD4(+) T cells to the lower airways. However, when the chronic lung disease develops fully (at 7 weeks after inoculation), it is driven instead by an innate immune axis that relies on invariant natural killer T (iNKT) cells that are programmed to activate macrophages to produce IL-13. The interaction between iNKT cells and macrophages depends on contact between the semi-invariant Valpha14Jalpha18-TCR on lung iNKT cells and the oligomorphic MHC-like protein CD1d on macrophages as well as NKT cell production of IL-13 that binds to the IL-13 receptor (IL-13R) on the macrophage. This innate immune axis is also activated in the lungs of humans with severe asthma or COPD based on detection of increased numbers of iNKT cells and alternatively activated IL-13-producing macrophages in the lung. Together, the findings identify an adaptive immune response that mediates acute disease and an innate immune response that drives chronic inflammatory lung disease in experimental and clinical settings.
Figures
Similar articles
-
New immune pathways from chronic post-viral lung disease.Ann N Y Acad Sci. 2010 Jan;1183(1):195-210. doi: 10.1111/j.1749-6632.2009.05136.x. Ann N Y Acad Sci. 2010. PMID: 20146716 Free PMC article. Review.
-
Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease.Nat Med. 2008 Jun;14(6):633-40. doi: 10.1038/nm1770. Epub 2008 May 18. Nat Med. 2008. PMID: 18488036 Free PMC article.
-
Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity.J Allergy Clin Immunol. 2012 Jan;129(1):216-27.e1-6. doi: 10.1016/j.jaci.2011.10.036. Epub 2011 Nov 25. J Allergy Clin Immunol. 2012. PMID: 22119406 Free PMC article.
-
Invariant NKT cells: regulation and function during viral infection.PLoS Pathog. 2012;8(8):e1002838. doi: 10.1371/journal.ppat.1002838. Epub 2012 Aug 16. PLoS Pathog. 2012. PMID: 22916008 Free PMC article. Review.
-
Ezh2 controls development of natural killer T cells, which cause spontaneous asthma-like pathology.J Allergy Clin Immunol. 2019 Aug;144(2):549-560.e10. doi: 10.1016/j.jaci.2019.02.024. Epub 2019 Mar 7. J Allergy Clin Immunol. 2019. PMID: 30851295
Cited by
-
The microbiome in asthma.J Allergy Clin Immunol. 2015 Jan;135(1):25-30. doi: 10.1016/j.jaci.2014.11.011. J Allergy Clin Immunol. 2015. PMID: 25567040 Free PMC article. Review.
-
Alternatively activated macrophages and airway disease.Chest. 2011 Sep;140(3):768-774. doi: 10.1378/chest.10-2132. Chest. 2011. PMID: 21896520 Free PMC article. Review.
-
Airway mucus function and dysfunction.N Engl J Med. 2010 Dec 2;363(23):2233-47. doi: 10.1056/NEJMra0910061. N Engl J Med. 2010. PMID: 21121836 Free PMC article. Review. No abstract available.
-
Perinatal and early childhood environmental factors influencing allergic asthma immunopathogenesis.Int Immunopharmacol. 2014 Sep;22(1):21-30. doi: 10.1016/j.intimp.2014.06.005. Epub 2014 Jun 19. Int Immunopharmacol. 2014. PMID: 24952205 Free PMC article. Review.
-
The role of early life viral bronchiolitis in the inception of asthma.Curr Opin Allergy Clin Immunol. 2013 Apr;13(2):211-6. doi: 10.1097/ACI.0b013e32835eb6ef. Curr Opin Allergy Clin Immunol. 2013. PMID: 23385289 Free PMC article. Review.
References
-
- Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, Dekruyff RH, Umetsu DH. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyper-reactivity. Nat Med. 2003;9:582–588. - PubMed
-
- Akbari O, Faul JL, Hoyte EG, Berry J, Wahlstrom J, Kronenberg M, Dekruyff RH, Umetsu DH. Cd4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N Engl J Med. 2006;354:1117–1129. - PubMed
-
- Anderson MS, Bluestone JA. The NOD mouse: A model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. - PubMed
-
- Bendelac A, Lantz O, Quimby ME, Yewdell JW, Bennink JR, Brutkiewicz RR. Cd1 recognition by mouse NKL+ T lymphocytes. Science. 1995;268:863–865. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
