

Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice
- PMID: 19477958
- PMCID: PMC2733819
- DOI: 10.1093/hmg/ddp253
Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice
Abstract
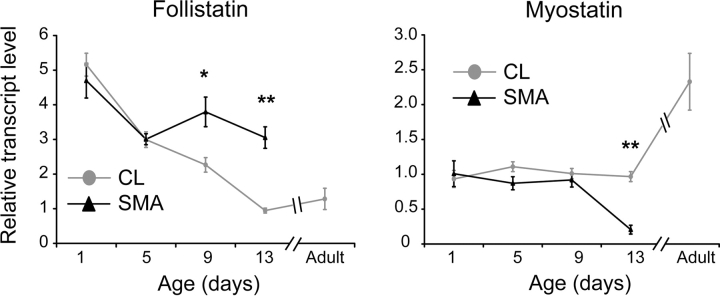
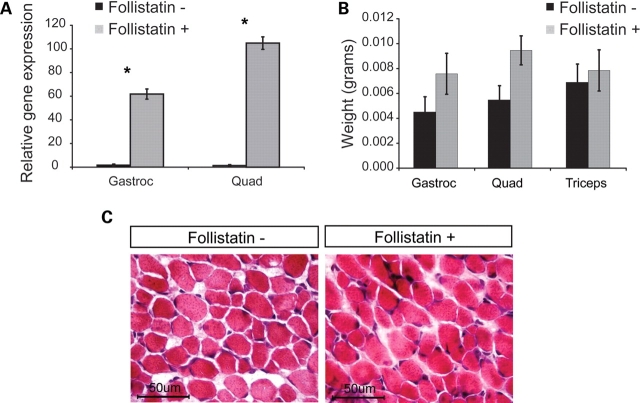
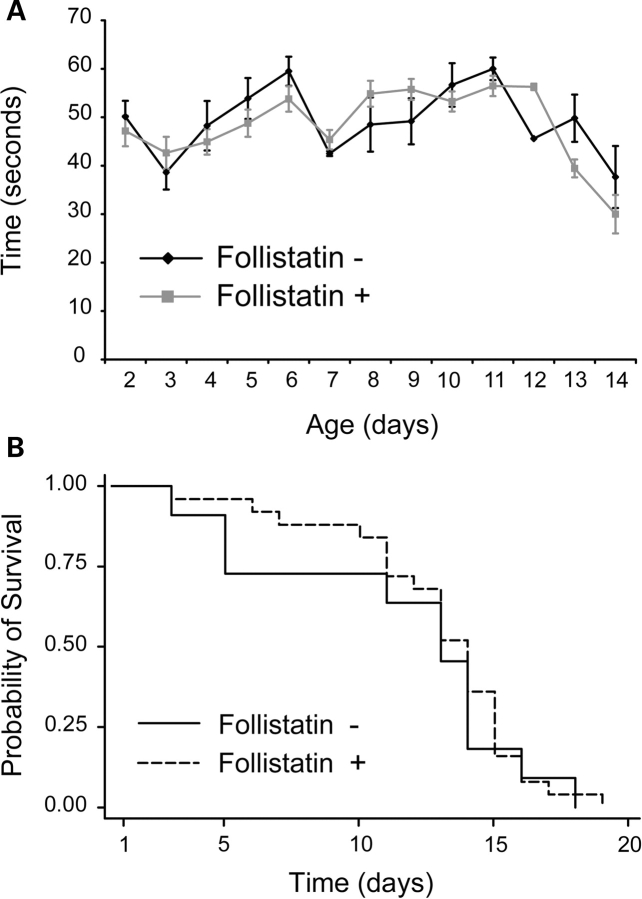
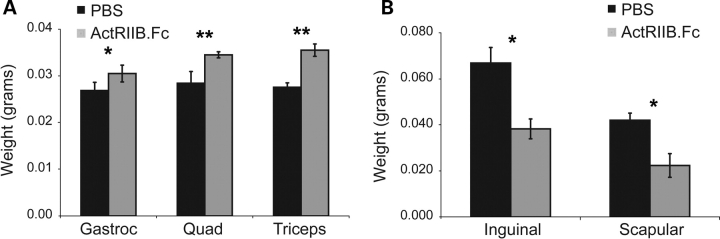
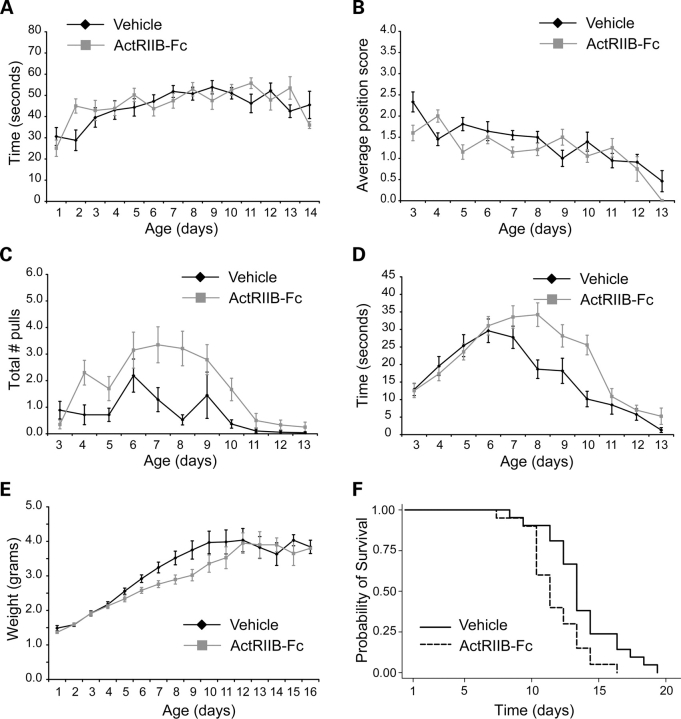
There is currently no treatment for the inherited motor neuron disease, spinal muscular atrophy (SMA). Severe SMA causes lower motor neuron loss, impaired myofiber development, profound muscle weakness and early mortality. Myostatin is a transforming growth factor-beta family member that inhibits muscle growth. Loss or blockade of myostatin signaling increases muscle mass and improves muscle strength in mouse models of primary muscle disease and in the motor neuron disease, amyotrophic lateral sclerosis. In this study, we evaluated the effects of blocking myostatin signaling in severe SMA mice (hSMN2/delta7SMN/mSmn(-/-)) by two independent strategies: (i) transgenic overexpression of the myostatin inhibitor follistatin and (ii) post-natal administration of a soluble activin receptor IIB (ActRIIB-Fc). SMA mice overexpressing follistatin showed little increase in muscle mass and no improvement in motor function or survival. SMA mice treated with ActRIIB-Fc showed minimal improvement in motor function, and no extension of survival compared with vehicle-treated mice. Together these results suggest that inhibition of myostatin may not be a promising therapeutic strategy in severe forms of SMA.
Figures
Similar articles
-
Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy.Neuromuscul Disord. 2012 Mar;22(3):277-85. doi: 10.1016/j.nmd.2011.10.012. Epub 2011 Nov 10. Neuromuscul Disord. 2012. PMID: 22079083
-
Activin Receptor Type IIB Inhibition Improves Muscle Phenotype and Function in a Mouse Model of Spinal Muscular Atrophy.PLoS One. 2016 Nov 21;11(11):e0166803. doi: 10.1371/journal.pone.0166803. eCollection 2016. PLoS One. 2016. PMID: 27870893 Free PMC article.
-
Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy.Hum Mol Genet. 2009 Mar 15;18(6):997-1005. doi: 10.1093/hmg/ddn426. Epub 2008 Dec 12. Hum Mol Genet. 2009. PMID: 19074460 Free PMC article.
-
Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases.Cell Mol Life Sci. 2022 Jun 21;79(7):374. doi: 10.1007/s00018-022-04408-w. Cell Mol Life Sci. 2022. PMID: 35727341 Free PMC article. Review.
-
Taldefgrobep Alfa and the Phase 3 RESILIENT Trial in Spinal Muscular Atrophy.Int J Mol Sci. 2024 Sep 24;25(19):10273. doi: 10.3390/ijms251910273. Int J Mol Sci. 2024. PMID: 39408601 Free PMC article. Review.
Cited by
-
Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders.Neurol Res Pract. 2022 Jan 4;4(1):2. doi: 10.1186/s42466-021-00162-9. Neurol Res Pract. 2022. PMID: 34983696 Free PMC article. Review.
-
Antisense oligonucleotides for the treatment of spinal muscular atrophy.Hum Gene Ther. 2013 May;24(5):489-98. doi: 10.1089/hum.2012.225. Hum Gene Ther. 2013. PMID: 23544870 Free PMC article. Review.
-
Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice.Am J Pathol. 2011 Feb;178(2):784-93. doi: 10.1016/j.ajpath.2010.10.035. Am J Pathol. 2011. PMID: 21281811 Free PMC article.
-
Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice.Hum Mol Genet. 2012 Oct 15;21(20):4448-59. doi: 10.1093/hmg/dds286. Epub 2012 Jul 13. Hum Mol Genet. 2012. PMID: 22798624 Free PMC article.
-
Dietary Implications of the Bidirectional Relationship between the Gut Microflora and Inflammatory Diseases with Special Emphasis on Irritable Bowel Disease: Current and Future Perspective.Nutrients. 2023 Jun 29;15(13):2956. doi: 10.3390/nu15132956. Nutrients. 2023. PMID: 37447285 Free PMC article. Review.
References
-
- Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G., Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 1997;16:265–269. - PubMed
-
- Dubowitz V. Muscle Disorders in Childhood. 2nd edn. Philadelphia: WB Saunders; 1995.
-
- Crawford T.O. Spinal muscular atrophies. In: Jones H., Vivo D.D., Darras B., editors. Neuromuscular Disorders of Infancy, Childhood and Adolescence: a Clinician's Approach. Philadephia: Butterworth Heinemann; 2003. pp. 145–166.
-
- Fidzianska A., Goebel H.H., Warlo I. Acute infantile spinal muscular atrophy. Muscle apoptosis as a proposed pathogenetic mechanism. Brain. 1990;113:433–445. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
