

Proteome coverage prediction with infinite Markov models
- PMID: 19477982
- PMCID: PMC2687987
- DOI: 10.1093/bioinformatics/btp233
Proteome coverage prediction with infinite Markov models
Abstract
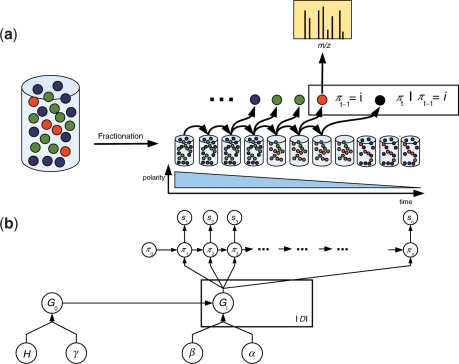
Motivation: Liquid chromatography tandem mass spectrometry (LC-MS/MS) is the predominant method to comprehensively characterize complex protein mixtures such as samples from prefractionated or complete proteomes. In order to maximize proteome coverage for the studied sample, i.e. identify as many traceable proteins as possible, LC-MS/MS experiments are typically repeated extensively and the results combined. Proteome coverage prediction is the task of estimating the number of peptide discoveries of future LC-MS/MS experiments. Proteome coverage prediction is important to enhance the design of efficient proteomics studies. To date, there does not exist any method to reliably estimate the increase of proteome coverage at an early stage.
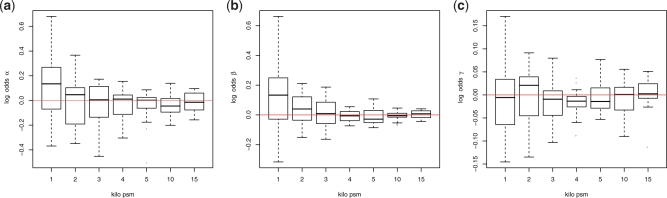
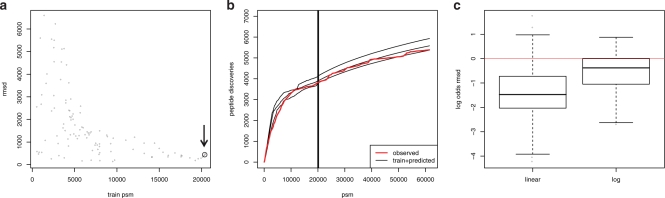
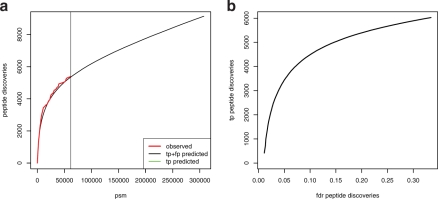
Results: We propose an extended infinite Markov model DiriSim to extrapolate the progression of proteome coverage based on a small number of already performed LC-MS/MS experiments. The method explicitly accounts for the uncertainty of peptide identifications. We tested DiriSim on a set of 37 LC-MS/MS experiments of a complete proteome sample and demonstrated that DiriSim correctly predicts the coverage progression already from a small subset of experiments. The predicted progression enabled us to specify maximal coverage for the test sample. We demonstrated that quality requirements on the final proteome map impose an upper bound on the number of useful experiment repetitions and limit the achievable proteome coverage.
Figures
References
-
- Antoniak CE. Mixtures of Dirichlet processes with applications to Bayesian nonparametric problems. Ann. Stat. 1974;2:1152–1174.
-
- Beal MJ, et al. Advances in Neural Information Processing Systems. Vol. 14. MIT Press; 2002. The infinite hidden Markov model.
-
- Blackwell D, MacQueen JB. Ferguson distributions via polya urn schemes. Ann. Stat. 1973;1:353–355.
-
- Brunner E, et al. A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 2007;25:576–583. - PubMed
-
- Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. - PubMed
