

Prediction of sub-cavity binding preferences using an adaptive physicochemical structure representation
- PMID: 19478002
- PMCID: PMC2687958
- DOI: 10.1093/bioinformatics/btp204
Prediction of sub-cavity binding preferences using an adaptive physicochemical structure representation
Abstract
Motivation: The ability to predict binding profiles for an arbitrary protein can significantly improve the areas of drug discovery, lead optimization and protein function prediction. At present, there are no successful algorithms capable of predicting binding profiles for novel proteins. Existing methods typically rely on manually curated templates or entire active site comparison. Consequently, they perform best when analyzing proteins sharing significant structural similarity with known proteins (i.e. proteins resulting from divergent evolution). These methods fall short when used to characterize the binding profile of a novel active site or one for which a template is not available. In contrast to previous approaches, our method characterizes the binding preferences of sub-cavities within the active site by exploiting a large set of known protein-ligand complexes. The uniqueness of our approach lies not only in the consideration of sub-cavities, but also in the more complete structural representation of these sub-cavities, their parametrization and the method by which they are compared. By only requiring local structural similarity, we are able to leverage previously unused structural information and perform binding inference for proteins that do not share significant structural similarity with known systems.
Results: Our algorithm demonstrates the ability to accurately cluster similar sub-cavities and to predict binding patterns across a diverse set of protein-ligand complexes. When applied to two high-profile drug targets, our algorithm successfully generates a binding profile that is consistent with known inhibitors. The results suggest that our algorithm should be useful in structure-based drug discovery and lead optimization.
Figures

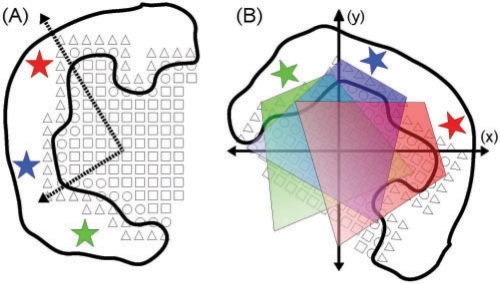
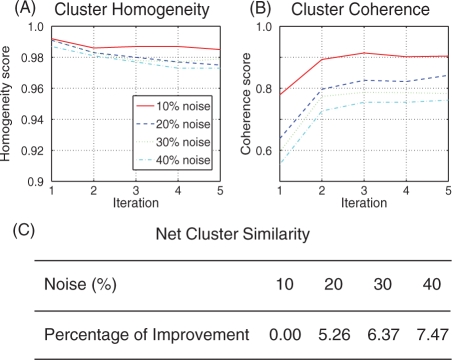
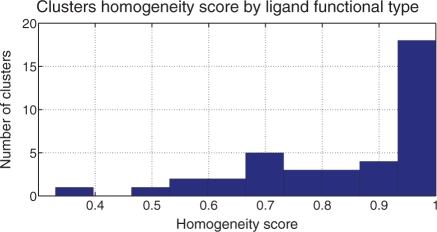
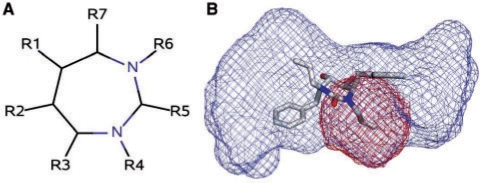
Similar articles
-
A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction.BMC Bioinformatics. 2010 Feb 22;11:99. doi: 10.1186/1471-2105-11-99. BMC Bioinformatics. 2010. PMID: 20175916 Free PMC article.
-
FINDSITE: a threading-based approach to ligand homology modeling.PLoS Comput Biol. 2009 Jun;5(6):e1000405. doi: 10.1371/journal.pcbi.1000405. Epub 2009 Jun 5. PLoS Comput Biol. 2009. PMID: 19503616 Free PMC article.
-
A unified statistical model to support local sequence order independent similarity searching for ligand-binding sites and its application to genome-based drug discovery.Bioinformatics. 2009 Jun 15;25(12):i305-12. doi: 10.1093/bioinformatics/btp220. Bioinformatics. 2009. PMID: 19478004 Free PMC article.
-
Chemogenomics in drug discovery: computational methods based on the comparison of binding sites.Future Med Chem. 2012 Oct;4(15):1971-9. doi: 10.4155/fmc.12.147. Future Med Chem. 2012. PMID: 23088277 Review.
-
Investigating drug-target association and dissociation mechanisms using metadynamics-based algorithms.Acc Chem Res. 2015 Feb 17;48(2):277-85. doi: 10.1021/ar500356n. Epub 2014 Dec 12. Acc Chem Res. 2015. PMID: 25496113 Review.
Cited by
-
Binding of protein kinase inhibitors to synapsin I inferred from pair-wise binding site similarity measurements.PLoS One. 2010 Aug 16;5(8):e12214. doi: 10.1371/journal.pone.0012214. PLoS One. 2010. PMID: 20808948 Free PMC article.
-
Considerations of Protein Subpockets in Fragment-Based Drug Design.Chem Biol Drug Des. 2016 Jan;87(1):5-20. doi: 10.1111/cbdd.12631. Epub 2015 Aug 31. Chem Biol Drug Des. 2016. PMID: 26307335 Free PMC article.
-
DrugOn: a fully integrated pharmacophore modeling and structure optimization toolkit.PeerJ. 2015 Jan 13;3:e725. doi: 10.7717/peerj.725. eCollection 2015. PeerJ. 2015. PMID: 25648563 Free PMC article.
-
Local functional descriptors for surface comparison based binding prediction.BMC Bioinformatics. 2012 Nov 24;13:314. doi: 10.1186/1471-2105-13-314. BMC Bioinformatics. 2012. PMID: 23176080 Free PMC article.
References
-
- Ala PJ, et al. Molecular recognition of cyclic urea HIV-1 protease inhibitors. J. Biol. Chem. 1998;273:12325–12331. - PubMed
-
- Binkowski AT, et al. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J. Mol. Biol. 2003;332:505–526. - PubMed
-
- Chen X, et al. Automated pharmacophore identification for large chemical data sets1. J. Chem. Infor. Comput. Sci. 1999;39:887–896. - PubMed
