

A unified statistical model to support local sequence order independent similarity searching for ligand-binding sites and its application to genome-based drug discovery
- PMID: 19478004
- PMCID: PMC2687974
- DOI: 10.1093/bioinformatics/btp220
A unified statistical model to support local sequence order independent similarity searching for ligand-binding sites and its application to genome-based drug discovery
Abstract
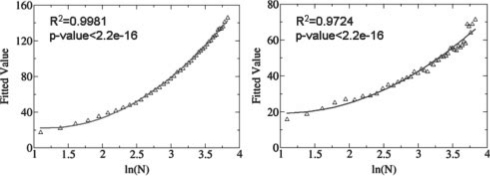
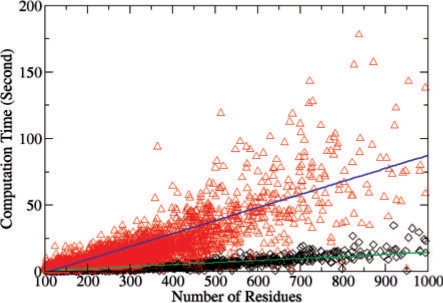
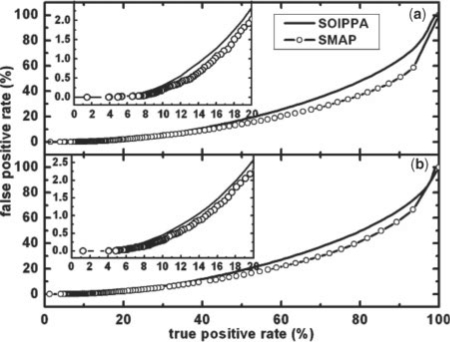
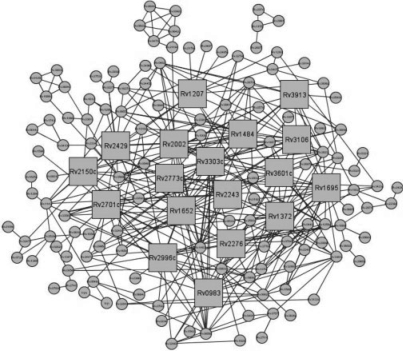
Functional relationships between proteins that do not share global structure similarity can be established by detecting their ligand-binding-site similarity. For a large-scale comparison, it is critical to accurately and efficiently assess the statistical significance of this similarity. Here, we report an efficient statistical model that supports local sequence order independent ligand-binding-site similarity searching. Most existing statistical models only take into account the matching vertices between two sites that are defined by a fixed number of points. In reality, the boundary of the binding site is not known or is dependent on the bound ligand making these approaches limited. To address these shortcomings and to perform binding-site mapping on a genome-wide scale, we developed a sequence-order independent profile-profile alignment (SOIPPA) algorithm that is able to detect local similarity between unknown binding sites a priori. The SOIPPA scoring integrates geometric, evolutionary and physical information into a unified framework. However, this imposes a significant challenge in assessing the statistical significance of the similarity because the conventional probability model that is based on fixed-point matching cannot be applied. Here we find that scores for binding-site matching by SOIPPA follow an extreme value distribution (EVD). Benchmark studies show that the EVD model performs at least two-orders faster and is more accurate than the non-parametric statistical method in the previous SOIPPA version. Efficient statistical analysis makes it possible to apply SOIPPA to genome-based drug discovery. Consequently, we have applied the approach to the structural genome of Mycobacterium tuberculosis to construct a protein-ligand interaction network. The network reveals highly connected proteins, which represent suitable targets for promiscuous drugs.
Figures

Similar articles
-
SMAP-WS: a parallel web service for structural proteome-wide ligand-binding site comparison.Nucleic Acids Res. 2010 Jul;38(Web Server issue):W441-4. doi: 10.1093/nar/gkq400. Epub 2010 May 19. Nucleic Acids Res. 2010. PMID: 20484373 Free PMC article.
-
Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments.Proc Natl Acad Sci U S A. 2008 Apr 8;105(14):5441-6. doi: 10.1073/pnas.0704422105. Epub 2008 Apr 2. Proc Natl Acad Sci U S A. 2008. PMID: 18385384 Free PMC article.
-
A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction.BMC Bioinformatics. 2010 Feb 22;11:99. doi: 10.1186/1471-2105-11-99. BMC Bioinformatics. 2010. PMID: 20175916 Free PMC article.
-
Computational methodologies for compound database searching that utilize experimental protein-ligand interaction information.Chem Biol Drug Des. 2010 Sep 1;76(3):191-200. doi: 10.1111/j.1747-0285.2010.01007.x. Epub 2010 Jul 15. Chem Biol Drug Des. 2010. PMID: 20636330 Review.
-
Chemogenomics in drug discovery: computational methods based on the comparison of binding sites.Future Med Chem. 2012 Oct;4(15):1971-9. doi: 10.4155/fmc.12.147. Future Med Chem. 2012. PMID: 23088277 Review.
Cited by
-
Protein function annotation by local binding site surface similarity.Proteins. 2014 Apr;82(4):679-94. doi: 10.1002/prot.24450. Epub 2013 Nov 22. Proteins. 2014. PMID: 24166661 Free PMC article.
-
Identification of distant drug off-targets by direct superposition of binding pocket surfaces.PLoS One. 2013 Dec 31;8(12):e83533. doi: 10.1371/journal.pone.0083533. eCollection 2013. PLoS One. 2013. PMID: 24391782 Free PMC article.
-
Rational discovery of dual-indication multi-target PDE/Kinase inhibitor for precision anti-cancer therapy using structural systems pharmacology.PLoS Comput Biol. 2019 Jun 17;15(6):e1006619. doi: 10.1371/journal.pcbi.1006619. eCollection 2019 Jun. PLoS Comput Biol. 2019. PMID: 31206508 Free PMC article.
-
VASP-E: specificity annotation with a volumetric analysis of electrostatic isopotentials.PLoS Comput Biol. 2014 Aug 28;10(8):e1003792. doi: 10.1371/journal.pcbi.1003792. eCollection 2014 Aug. PLoS Comput Biol. 2014. PMID: 25166865 Free PMC article.
-
eMatchSite: sequence order-independent structure alignments of ligand binding pockets in protein models.PLoS Comput Biol. 2014 Sep 18;10(9):e1003829. doi: 10.1371/journal.pcbi.1003829. eCollection 2014 Sep. PLoS Comput Biol. 2014. PMID: 25232727 Free PMC article.
References
-
- Andreeva A, Murzin AG. Evolution of protein fold in the presence of functional constraints. Curr. Opin. Struct. Biol. 2006;16:399–408. - PubMed
-
- Artymiuk PJ, et al. A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side-chains in protein structures. J. Mol. Biol. 1994;243:327–344. - PubMed
-
- Barker JA, Thornton JM. An algorithm for constraint-based structural template matching: application to 3D templates with statistical analysis. Bioinformatics. 2003;19:1644–1649. - PubMed
-
- Bashton M, Chothia C. The generation of new protein functions by the combination of domains. Structure. 2007;15:85–99. - PubMed
