

Identifying novel constrained elements by exploiting biased substitution patterns
- PMID: 19478016
- PMCID: PMC2687944
- DOI: 10.1093/bioinformatics/btp190
Identifying novel constrained elements by exploiting biased substitution patterns
Abstract
Motivation: Comparing the genomes from closely related species provides a powerful tool to identify functional elements in a reference genome. Many methods have been developed to identify conserved sequences across species; however, existing methods only model conservation as a decrease in the rate of mutation and have ignored selection acting on the pattern of mutations.
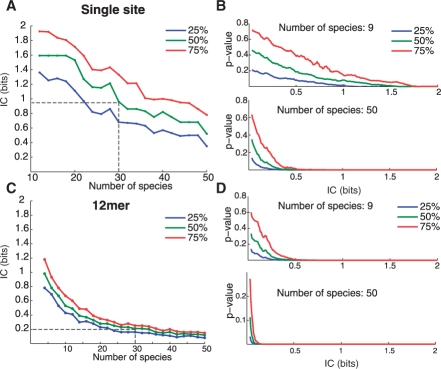
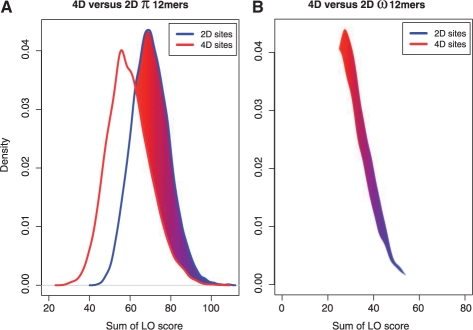
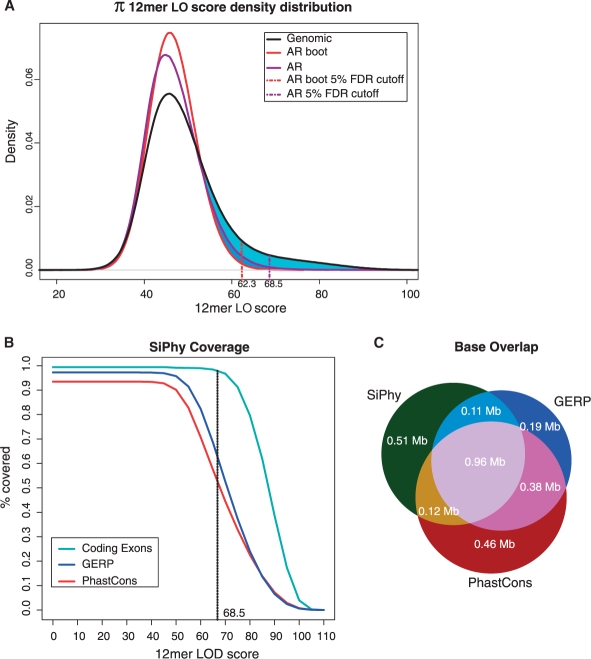
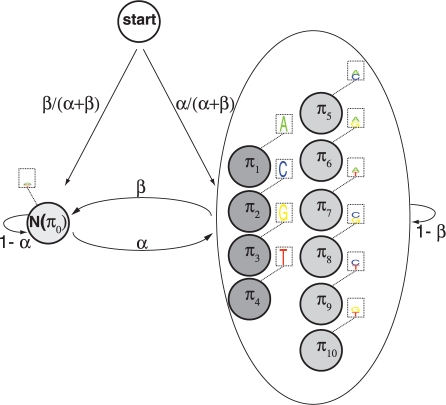
Results: We present a new approach that takes advantage of deeply sequenced clades to identify evolutionary selection by uncovering not only signatures of rate-based conservation but also substitution patterns characteristic of sequence undergoing natural selection. We describe a new statistical method for modeling biased nucleotide substitutions, a learning algorithm for inferring site-specific substitution biases directly from sequence alignments and a hidden Markov model for detecting constrained elements characterized by biased substitutions. We show that the new approach can identify significantly more degenerate constrained sequences than rate-based methods. Applying it to the ENCODE regions, we identify as much as 10.2% of these regions are under selection.
Availability: The algorithms are implemented in a Java software package, called SiPhy, freely available at http://www.broadinstitute.org/science/software/.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Bejerano G, et al. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006;441:87–90. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
