

Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing
- PMID: 19478811
- PMCID: PMC2990409
- DOI: 10.1038/nprot.2009.68
Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing
Abstract
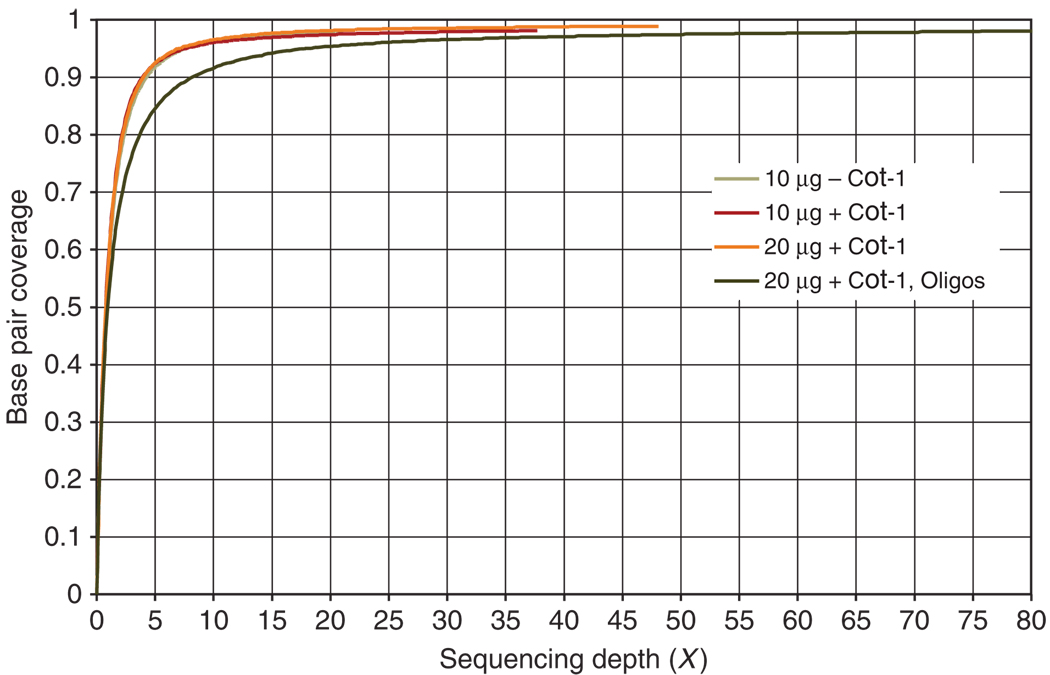
Complementary techniques that deepen information content and minimize reagent costs are required to realize the full potential of massively parallel sequencing. Here, we describe a resequencing approach that directs focus to genomic regions of high interest by combining hybridization-based purification of multi-megabase regions with sequencing on the Illumina Genome Analyzer (GA). The capture matrix is created by a microarray on which probes can be programmed as desired to target any non-repeat portion of the genome, while the method requires only a basic familiarity with microarray hybridization. We present a detailed protocol suitable for 1-2 microg of input genomic DNA and highlight key design tips in which high specificity (>65% of reads stem from enriched exons) and high sensitivity (98% targeted base pair coverage) can be achieved. We have successfully applied this to the enrichment of coding regions, in both human and mouse, ranging from 0.5 to 4 Mb in length. From genomic DNA library production to base-called sequences, this procedure takes approximately 9-10 d inclusive of array captures and one Illumina flow cell run.
Figures
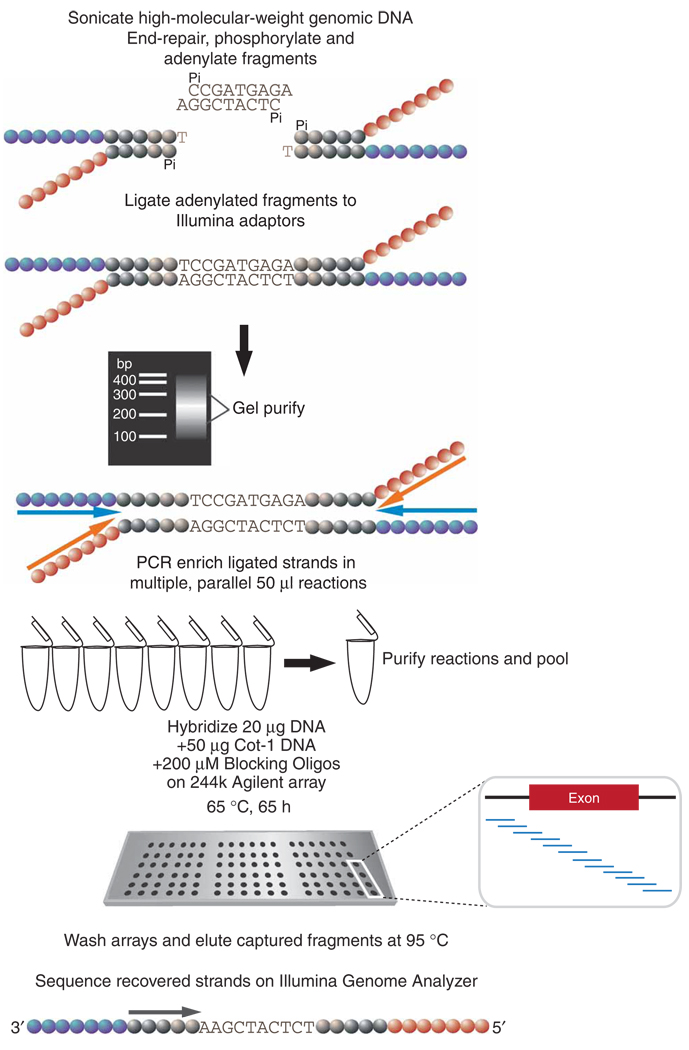
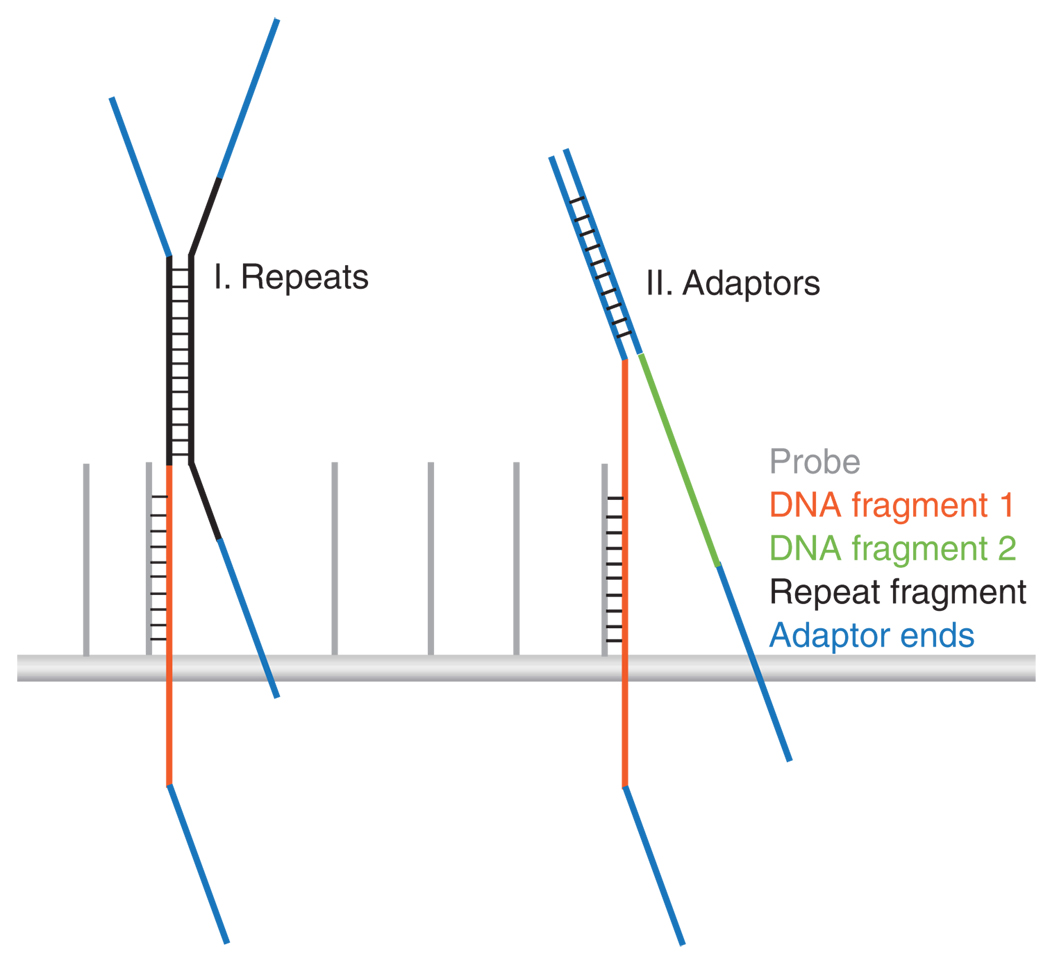
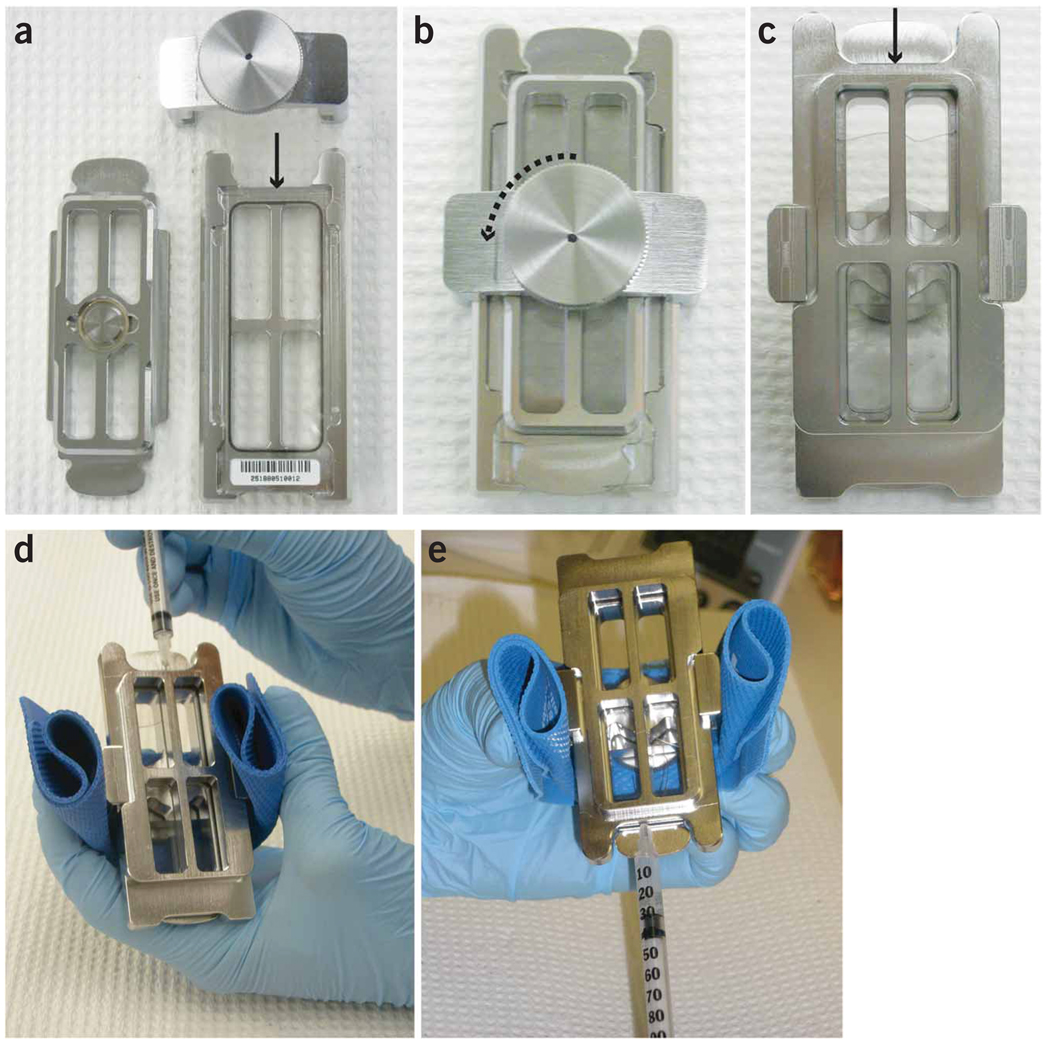
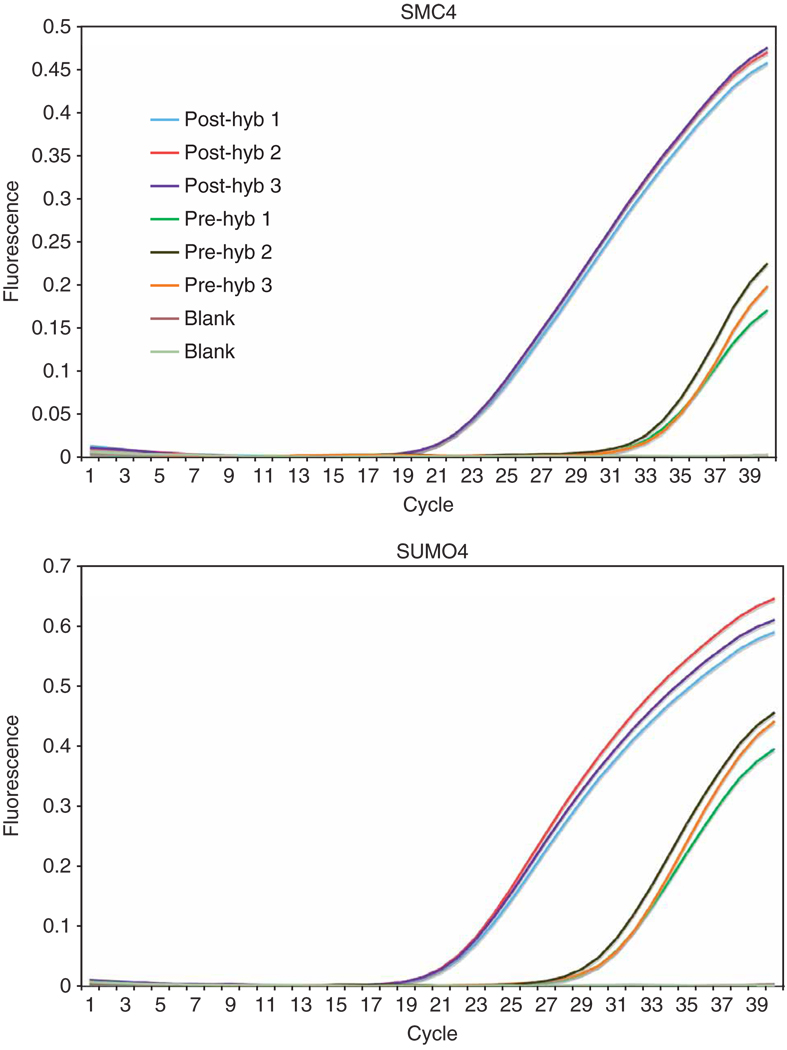
References
-
- Kaiser J. DNA sequencing. A plan to capture human diversity in 1000 genomes. Science. 2008;319:395. - PubMed
-
- Siva N. 1000 Genomes Project. Nat. Biotechnol. 2008;26:256. - PubMed
-
- Collins FS, Barker AD. Mapping the cancer genome. Pinpointing the genes involved in cancer will help chart anew course across the complex landscape of human malignancies. Sci. Am. 2007;296:50–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
