

Viral sequestration of antigen subverts cross presentation to CD8(+) T cells
- PMID: 19478869
- PMCID: PMC2680035
- DOI: 10.1371/journal.ppat.1000457
Viral sequestration of antigen subverts cross presentation to CD8(+) T cells
Abstract
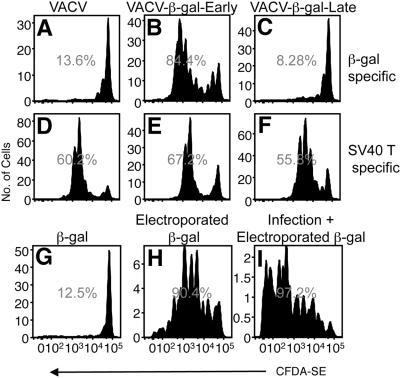
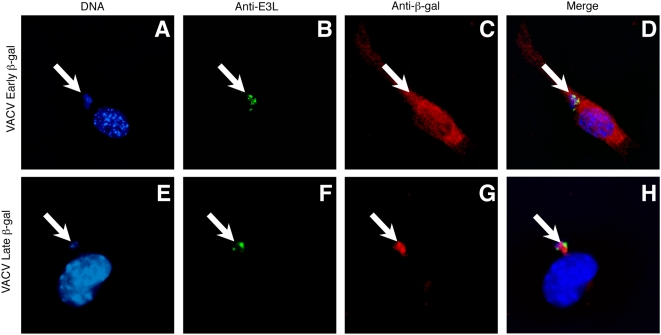
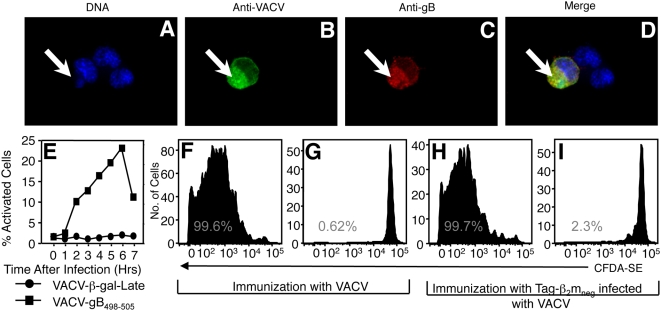
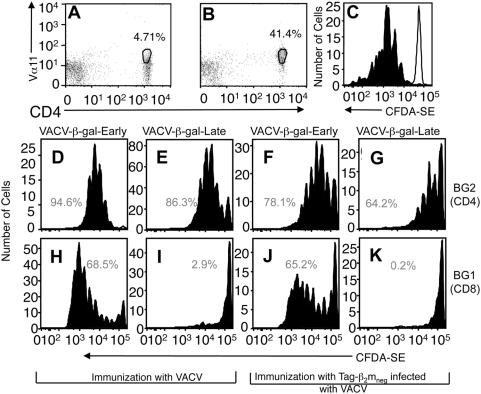
Virus-specific CD8(+) T cells (T(CD8+)) are initially triggered by peptide-MHC Class I complexes on the surface of professional antigen presenting cells (pAPC). Peptide-MHC complexes are produced by two spatially distinct pathways during virus infection. Endogenous antigens synthesized within virus-infected pAPC are presented via the direct-presentation pathway. Many viruses have developed strategies to subvert direct presentation. When direct presentation is blocked, the cross-presentation pathway, in which antigen is transferred from virus-infected cells to uninfected pAPC, is thought to compensate and allow the generation of effector T(CD8+). Direct presentation of vaccinia virus (VACV) antigens driven by late promoters does not occur, as an abortive infection of pAPC prevents production of these late antigens. This lack of direct presentation results in a greatly diminished or ablated T(CD8+) response to late antigens. We demonstrate that late poxvirus antigens do not enter the cross-presentation pathway, even when identical antigens driven by early promoters access this pathway efficiently. The mechanism mediating this novel means of viral modulation of antigen presentation involves the sequestration of late antigens within virus factories. Early antigens and cellular antigens are cross-presented from virus-infected cells, as are late antigens that are targeted to compartments outside of the virus factories. This virus-mediated blockade specifically targets the cross-presentation pathway, since late antigen that is not cross-presented efficiently enters the MHC Class II presentation pathway. These data are the first to describe an evasion mechanism employed by pathogens to prevent entry into the cross-presentation pathway. In the absence of direct presentation, this evasion mechanism leads to a complete ablation of the T(CD8+) response and a potential replicative advantage for the virus. Such mechanisms of viral modulation of antigen presentation must also be taken into account during the rational design of antiviral vaccines.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Peptide-MHC-I from Endogenous Antigen Outnumber Those from Exogenous Antigen, Irrespective of APC Phenotype or Activation.PLoS Pathog. 2015 Jun 24;11(6):e1004941. doi: 10.1371/journal.ppat.1004941. eCollection 2015 Jun. PLoS Pathog. 2015. PMID: 26107264 Free PMC article.
-
Prolonged antigen presentation following an acute virus infection requires direct and then cross-presentation.J Immunol. 2014 Oct 15;193(8):4169-77. doi: 10.4049/jimmunol.1302565. Epub 2014 Sep 15. J Immunol. 2014. PMID: 25225666 Free PMC article.
-
Influenza A virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells.PLoS Pathog. 2012;8(3):e1002572. doi: 10.1371/journal.ppat.1002572. Epub 2012 Mar 8. PLoS Pathog. 2012. PMID: 22412374 Free PMC article.
-
The cross-priming pathway: a portrait of an intricate immune system.Scand J Immunol. 2007 Apr;65(4):311-9. doi: 10.1111/j.1365-3083.2007.01909.x. Scand J Immunol. 2007. PMID: 17386021 Review.
-
Cross-presentation: underlying mechanisms and role in immune surveillance.Immunol Rev. 2005 Oct;207:166-83. doi: 10.1111/j.0105-2896.2005.00301.x. Immunol Rev. 2005. PMID: 16181335 Review.
Cited by
-
Is The Allergen Really Needed in Allergy Immunotherapy?Curr Treat Options Allergy. 2015;2(1):72-82. doi: 10.1007/s40521-014-0038-5. Curr Treat Options Allergy. 2015. PMID: 25722959 Free PMC article. Review.
-
Regulation of CD8(+) T Cell Responses to Retinal Antigen by Local FoxP3(+) Regulatory T Cells.Front Immunol. 2012 Jun 21;3:166. doi: 10.3389/fimmu.2012.00166. eCollection 2012. Front Immunol. 2012. PMID: 22737153 Free PMC article.
-
Efficient Induction of Cytotoxic T Cells by Viral Vector Vaccination Requires STING-Dependent DC Functions.Front Immunol. 2020 Jul 16;11:1458. doi: 10.3389/fimmu.2020.01458. eCollection 2020. Front Immunol. 2020. PMID: 32765505 Free PMC article.
-
Sequestration of Late Antigens Within Viral Factories Impairs MVA Vector-Induced Protective Memory CTL Responses.Front Immunol. 2019 Dec 4;10:2850. doi: 10.3389/fimmu.2019.02850. eCollection 2019. Front Immunol. 2019. PMID: 31867011 Free PMC article.
-
Cross-priming in health and disease.Nat Rev Immunol. 2010 Jun;10(6):403-14. doi: 10.1038/nri2780. Nat Rev Immunol. 2010. PMID: 20498667 Review.
References
-
- Yewdell JW, Norbury CC, Bennink JR. Mechanisms of exogenous antigen presentation by MHC class I molecules in vitro and in vivo: implications for generating CD8+ T cell responses to infectious agents, tumors, transplants, and vaccines. Adv Immunol. 1999;73:1–77. - PubMed
-
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, et al. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. - PubMed
-
- Norbury CC, Basta S, Donohue KB, Tscharke DC, Princiotta MF, et al. CD8+ T cell cross-priming via transfer of proteasome substrates. Science. 2004;304:1318–1321. - PubMed
-
- Norbury CC, Sigal LJ. Cross priming or direct priming: is that really the question? Curr Opin Immunol. 2003;15:82–88. - PubMed
-
- Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3:1019–1025. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
