

Comparative genomic characterization of Francisella tularensis strains belonging to low and high virulence subspecies
- PMID: 19478886
- PMCID: PMC2682660
- DOI: 10.1371/journal.ppat.1000459
Comparative genomic characterization of Francisella tularensis strains belonging to low and high virulence subspecies
Abstract
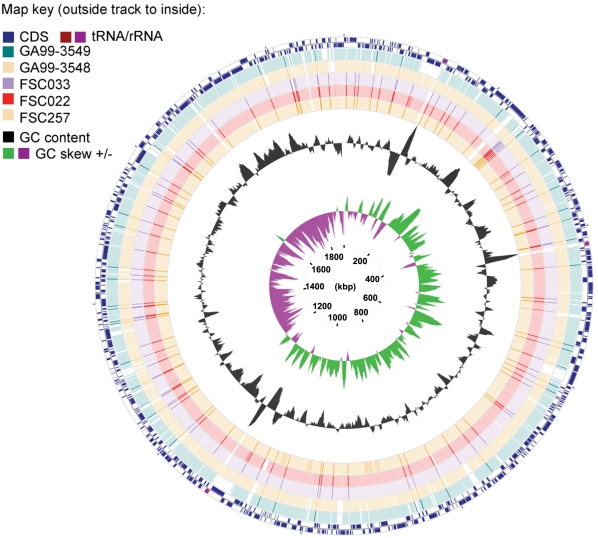
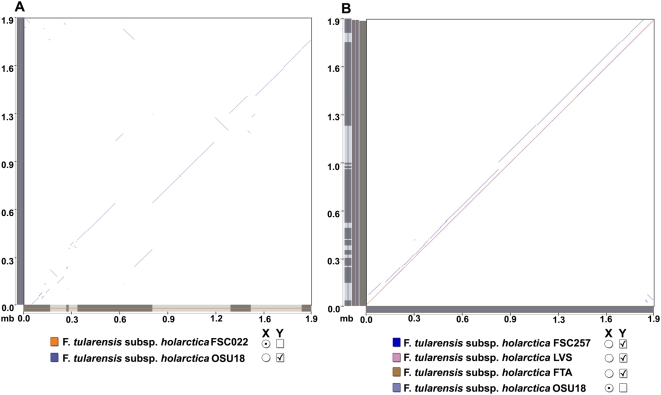
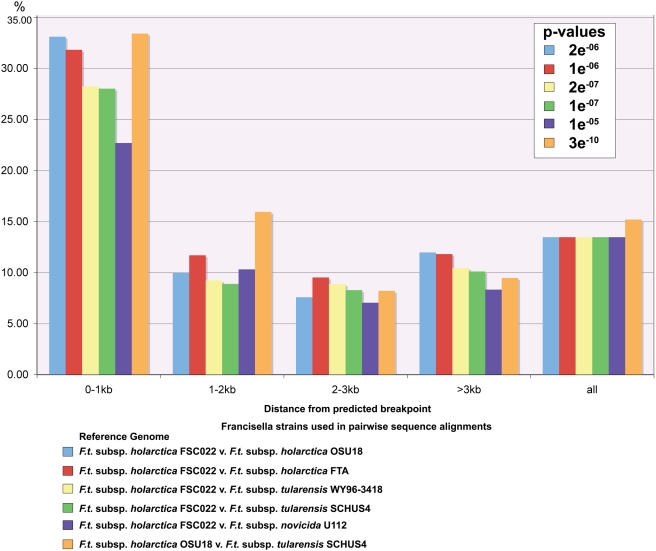
Tularemia is a geographically widespread, severely debilitating, and occasionally lethal disease in humans. It is caused by infection by a gram-negative bacterium, Francisella tularensis. In order to better understand its potency as an etiological agent as well as its potential as a biological weapon, we have completed draft assemblies and report the first complete genomic characterization of five strains belonging to the following different Francisella subspecies (subsp.): the F. tularensis subsp. tularensis FSC033, F. tularensis subsp. holarctica FSC257 and FSC022, and F. tularensis subsp. novicida GA99-3548 and GA99-3549 strains. Here, we report the sequencing of these strains and comparative genomic analysis with recently available public Francisella sequences, including the rare F. tularensis subsp. mediasiatica FSC147 strain isolate from the Central Asian Region. We report evidence for the occurrence of large-scale rearrangement events in strains of the holarctica subspecies, supporting previous proposals that further phylogenetic subdivisions of the Type B clade are likely. We also find a significant enrichment of disrupted or absent ORFs proximal to predicted breakpoints in the FSC022 strain, including a genetic component of the Type I restriction-modification defense system. Many of the pseudogenes identified are also disrupted in the closely related rarely human pathogenic F. tularensis subsp. mediasiatica FSC147 strain, including modulator of drug activity B (mdaB) (FTT0961), which encodes a known NADPH quinone reductase involved in oxidative stress resistance. We have also identified genes exhibiting sequence similarity to effectors of the Type III (T3SS) and components of the Type IV secretion systems (T4SS). One of the genes, msrA2 (FTT1797c), is disrupted in F. tularensis subsp. mediasiatica and has recently been shown to mediate bacterial pathogen survival in host organisms. Our findings suggest that in addition to the duplication of the Francisella Pathogenicity Island, and acquisition of individual loci, adaptation by gene loss in the more recently emerged tularensis, holarctica, and mediasiatica subspecies occurred and was distinct from evolutionary events that differentiated these subspecies, and the novicida subspecies, from a common ancestor. Our findings are applicable to future studies focused on variations in Francisella subspecies pathogenesis, and of broader interest to studies of genomic pathoadaptation in bacteria.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- CDC. 2008. Tularemia http://www.cdc.gov/ncidod/diseases/submenus/sub_tularemia.htm.
-
- Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
