

Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia
- PMID: 19481194
- PMCID: PMC2694977
- DOI: 10.1016/j.ajhg.2009.05.002
Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia
Abstract
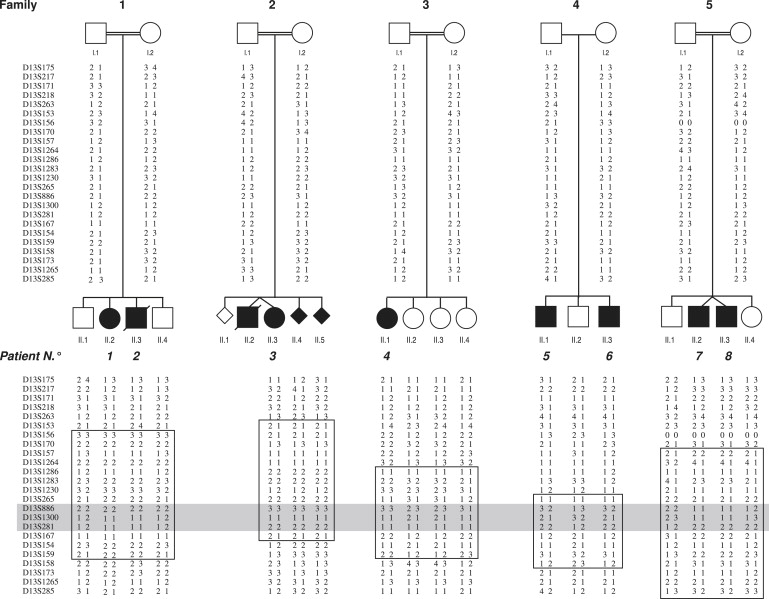
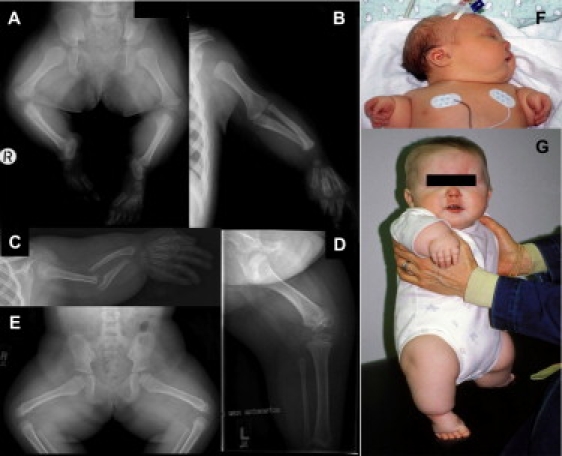
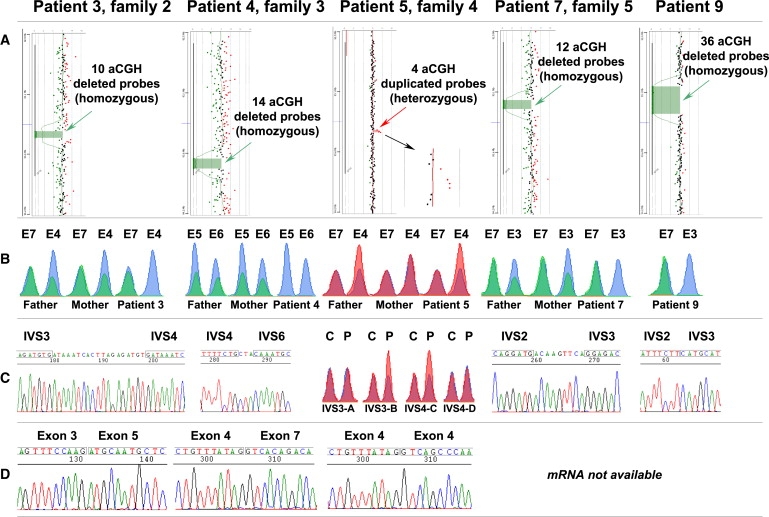
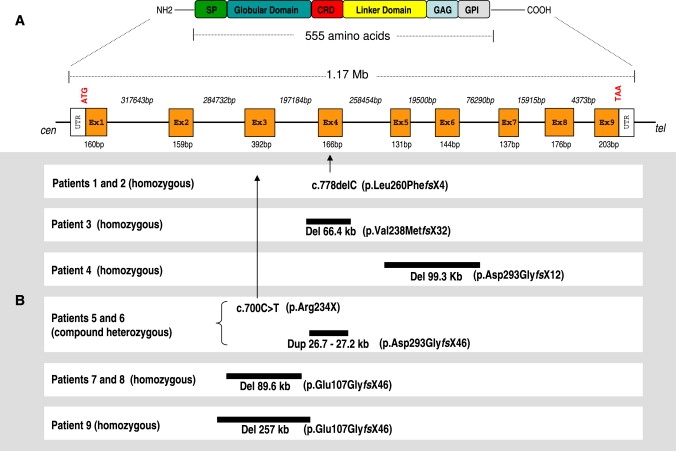
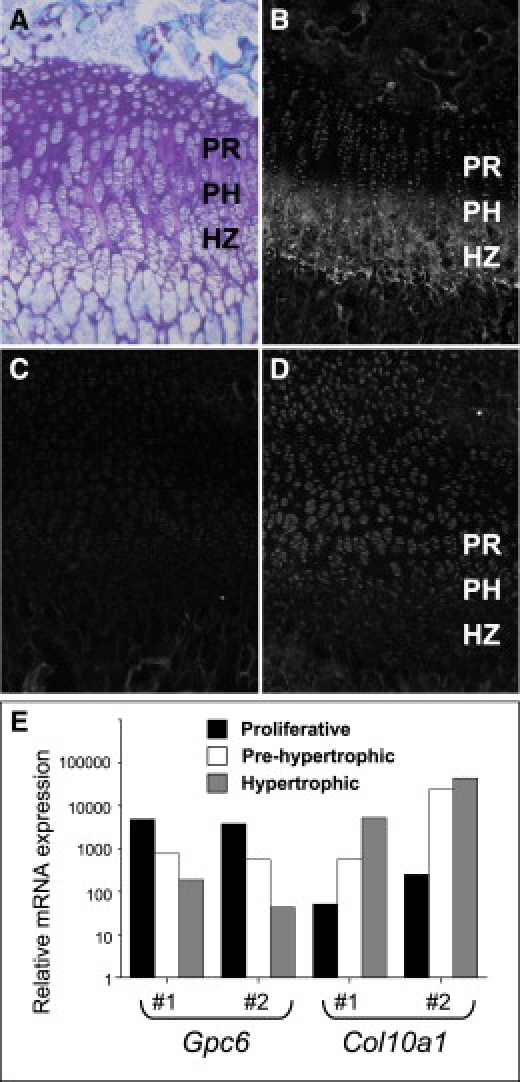
Glypicans are a family of glycosylphosphatidylinositol (GPI)-anchored, membrane-bound heparan sulfate (HS) proteoglycans. Their biological roles are only partly understood, although it is assumed that they modulate the activity of HS-binding growth factors. The involvement of glypicans in developmental morphogenesis and growth regulation has been highlighted by Drosophila mutants and by a human overgrowth syndrome with multiple malformations caused by glypican 3 mutations (Simpson-Golabi-Behmel syndrome). We now report that autosomal-recessive omodysplasia, a genetic condition characterized by short-limbed short stature, craniofacial dysmorphism, and variable developmental delay, maps to chromosome 13 (13q31.1-q32.2) and is caused by point mutations or by larger genomic rearrangements in glypican 6 (GPC6). All mutations cause truncation of the GPC6 protein and abolish both the HS-binding site and the GPI-bearing membrane-associated domain, and thus loss of function is predicted. Expression studies in microdissected mouse growth plate revealed expression of Gpc6 in proliferative chondrocytes. Thus, GPC6 seems to have a previously unsuspected role in endochondral ossification and skeletal growth, and its functional abrogation results in a short-limb phenotype.
Figures
References
-
- Kirkpatrick C.A., Knox S.M., Staatz W.D., Fox B., Lercher D.M., Selleck S.B. The function of a Drosophila glypican does not depend entirely on heparan sulfate modification. Dev. Biol. 2006;300:570–582. - PubMed
-
- Filmus J. Glypicans in growth control and cancer. Glycobiology. 2001;11:19R–23R. - PubMed
-
- Nakato H., Kimata K. Heparan sulfate fine structure and specificity of proteoglycan functions. Biochim. Biophys. Acta. 2002;1573:312–318. - PubMed
-
- Filmus J. The contribution of in vivo manipulation of gene expression to the understanding of the function of glypicans. Glycoconj. J. 2002;19:319–323. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
