

Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells
- PMID: 19487416
- PMCID: PMC2715069
- DOI: 10.1084/jem.20090299
Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells
Abstract
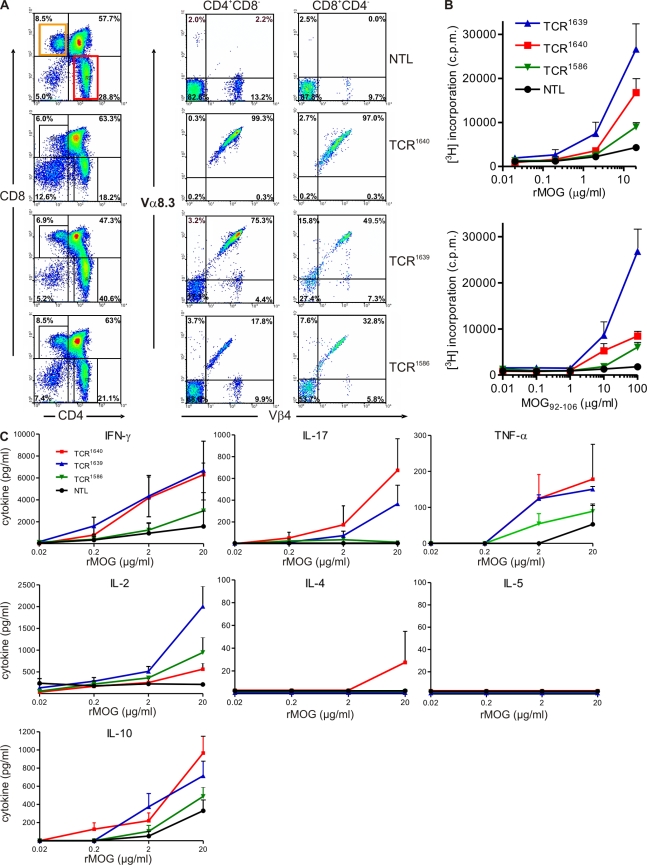
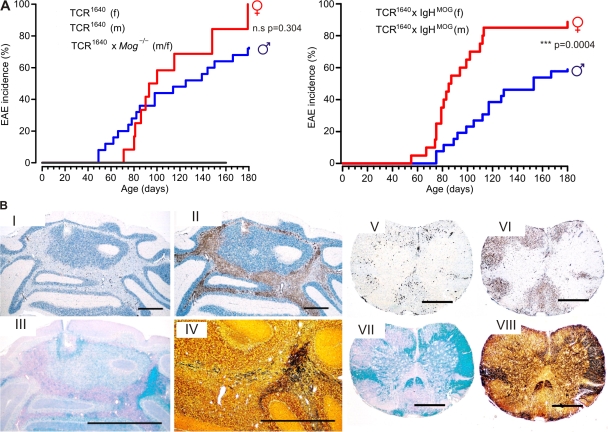
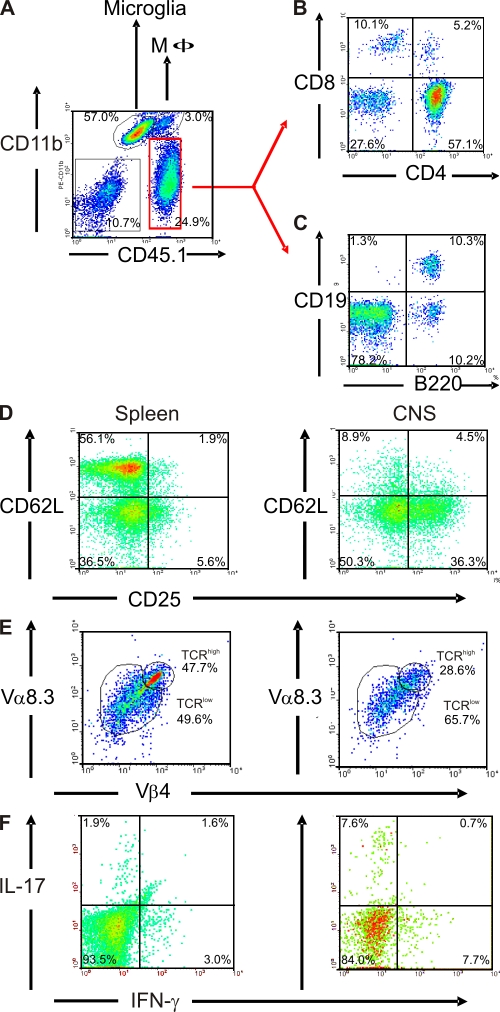
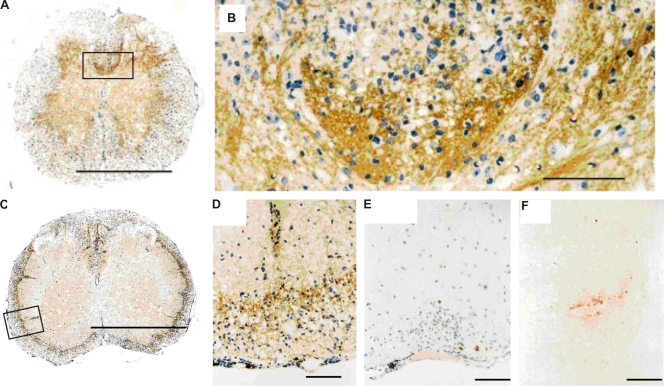
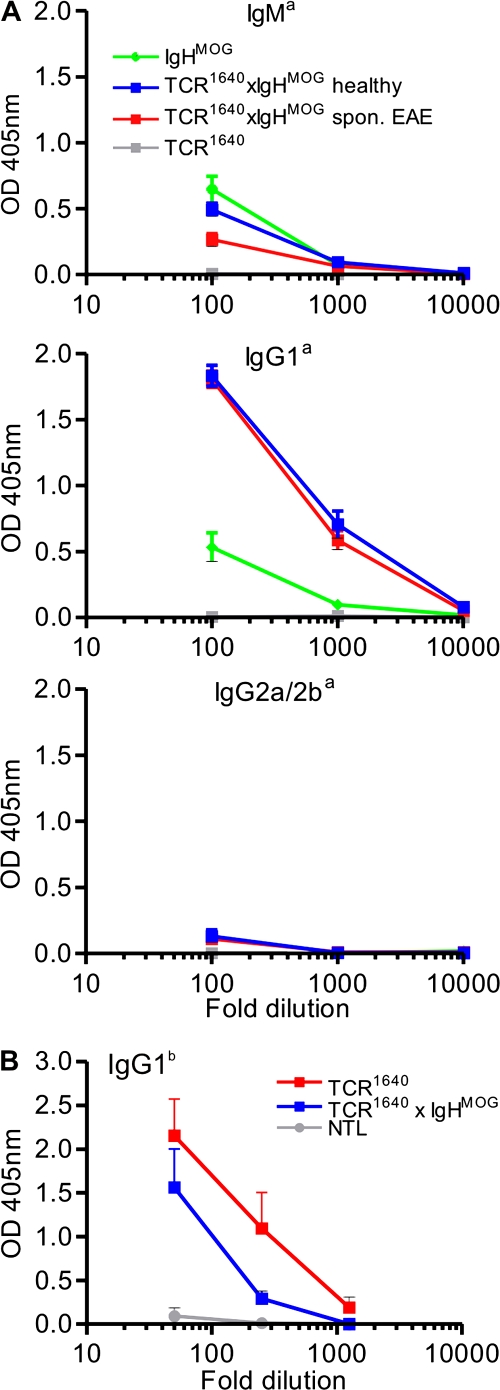
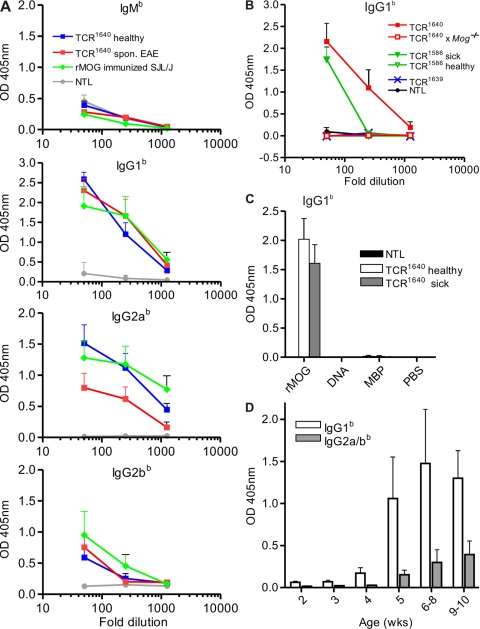
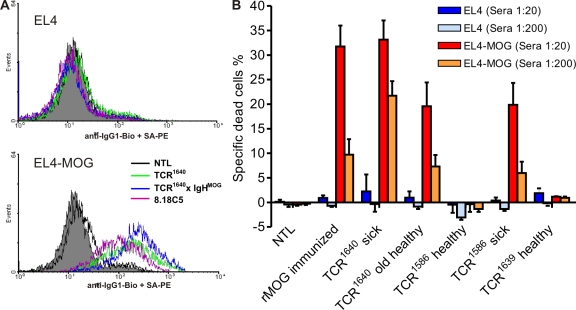
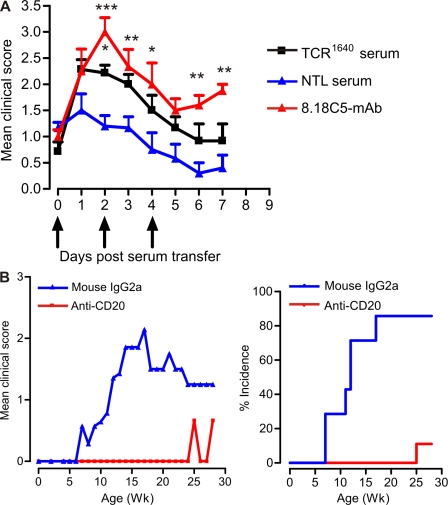
We describe new T cell receptor (TCR) transgenic mice (relapsing-remitting [RR] mice) carrying a TCR specific for myelin oligodendrocyte glycoprotein (MOG) peptide 92-106 in the context of I-A(s). Backcrossed to the SJL/J background, most RR mice spontaneously develop RR experimental autoimmune encephalomyelitis (EAE) with episodes often altering between different central nervous system tissues like the cerebellum, optic nerve, and spinal cord. Development of spontaneous EAE depends on the presence of an intact B cell compartment and on the expression of MOG autoantigen. There is no spontaneous EAE development in B cell-depleted mice or in transgenic mice lacking MOG. Transgenic T cells seem to expand MOG autoreactive B cells from the endogenous repertoire. The expanded autoreactive B cells produce autoantibodies binding to a conformational epitope on the native MOG protein while ignoring the T cell target peptide. The secreted autoantibodies are pathogenic, enhancing demyelinating EAE episodes. RR mice constitute the first spontaneous animal model for the most common form of multiple sclerosis (MS), RR MS.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
