

Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome
- PMID: 19487419
- PMCID: PMC2715068
- DOI: 10.1084/jem.20082767
Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome
Abstract
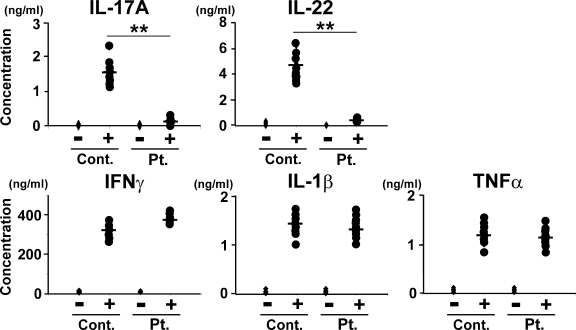
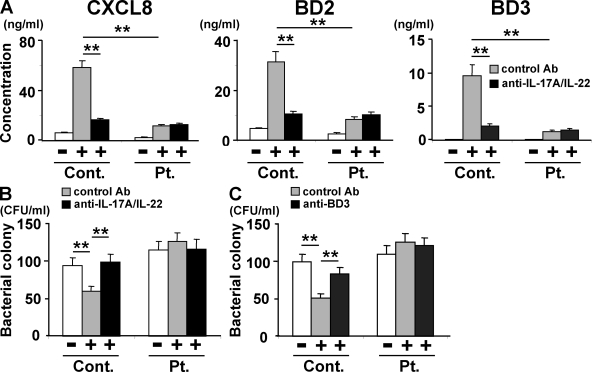
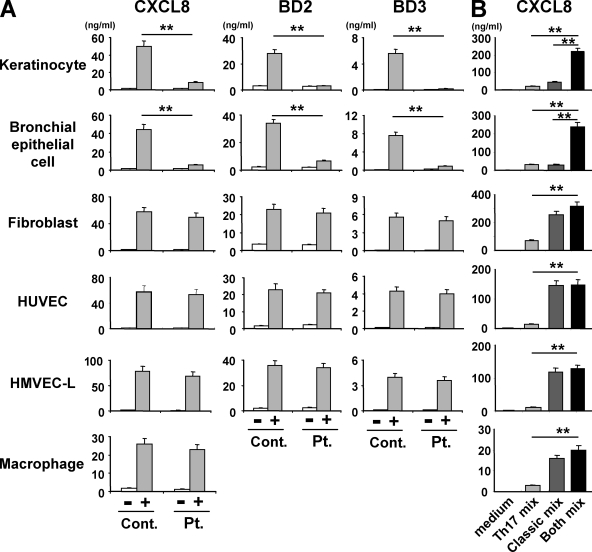
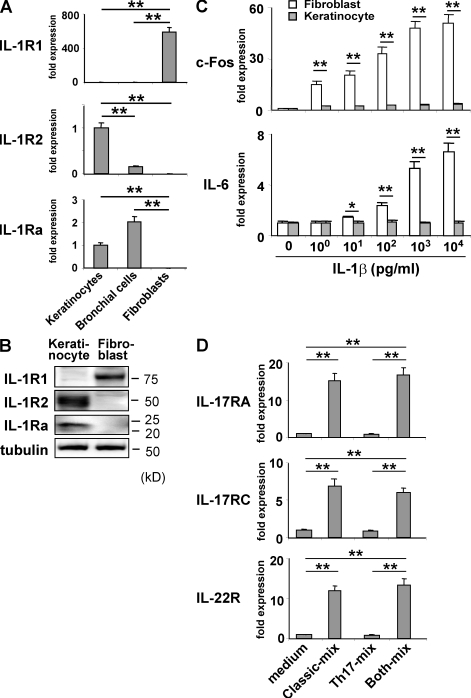
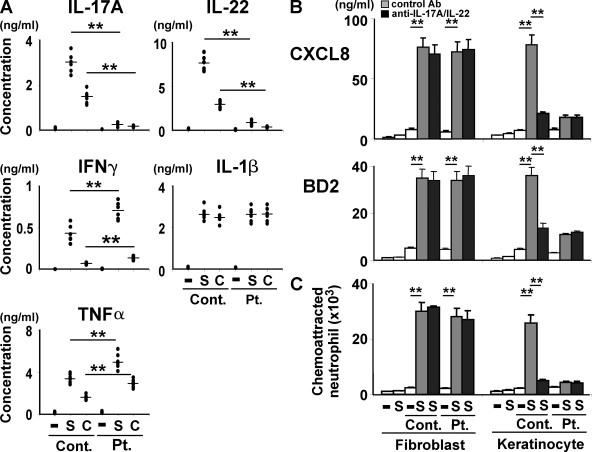
Hyper-IgE syndrome (HIES) is a primary immunodeficiency characterized by atopic manifestations and susceptibility to infections with extracellular pathogens, typically Staphylococcus aureus, which preferentially affect the skin and lung. Previous studies reported the defective differentiation of T helper 17 (Th17) cells in HIES patients caused by hypomorphic STAT3 mutations. However, the apparent contradiction between the systemic Th17 deficiency and the skin/lung-restricted susceptibility to staphylococcal infections remains puzzling. We present a possible molecular explanation for this enigmatic contradiction. HIES T cells showed impaired production of Th17 cytokines but normal production of classical proinflammatory cytokines including interleukin 1beta. Normal human keratinocytes and bronchial epithelial cells were deeply dependent on the synergistic action of Th17 cytokines and classical proinflammatory cytokines for their production of antistaphylococcal factors, including neutrophil-recruiting chemokines and antimicrobial peptides. In contrast, other cell types were efficiently stimulated with the classical proinflammatory cytokines alone to produce such factors. Accordingly, keratinocytes and bronchial epithelial cells, unlike other cell types, failed to produce antistaphylococcal factors in response to HIES T cell-derived cytokines. These results appear to explain, at least in part, why HIES patients suffer from recurrent staphylococcal infections confined to the skin and lung in contrast to more systemic infections in neutrophil-deficient patients.
Figures
References
-
- Dong C. 2008. TH17 cells in development: an updated view of their molecular identity and genetic programming.Nat. Rev. Immunol. 8:337–348 - PubMed
-
- McGeachy M.J., Bak-Jensen K.S., Chen Y., Tato C.M., Blumenschein W., McClanahan T., Cua D.J. 2007. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology.Nat. Immunol. 8:1390–1397 - PubMed
-
- Cua D.J., Sherlock J., Chen Y., Murphy C.A., Joyce B., Seymour B., Lucian L., To W., Kwan S., Churakova T., et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain.Nature. 421:744–748 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
