

Kinetic control of negative feedback regulators of NF-kappaB/RelA determines their pathogen- and cytokine-receptor signaling specificity
- PMID: 19487661
- PMCID: PMC2701028
- DOI: 10.1073/pnas.0812367106
Kinetic control of negative feedback regulators of NF-kappaB/RelA determines their pathogen- and cytokine-receptor signaling specificity
Abstract
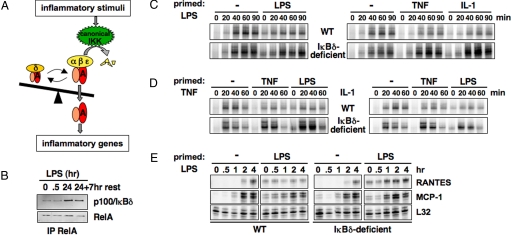
Mammalian signaling networks contain an abundance of negative feedback regulators that may have overlapping ("fail-safe") or specific functions. Within the NF-kappaB signaling module, IkappaB alpha is known as a negative feedback regulator, but the newly characterized inhibitor IkappaB delta is also inducibly expressed in response to inflammatory stimuli. To examine IkappaB delta's roles in inflammatory signaling, we mathematically modeled the 4-IkappaB-containing NF-kappaB signaling module and developed a computational phenotyping methodology of general applicability. We found that IkappaB delta, like IkappaB alpha, can provide negative feedback, but each functions stimulus-specifically. Whereas IkappaB delta attenuates persistent, pathogen-triggered signals mediated by TLRs, the more prominent IkappaB alpha does not. Instead, IkappaB alpha, which functions more rapidly, is primarily involved in determining the temporal profile of NF-kappaB signaling in response to cytokines that serve intercellular communication. Indeed, when removing the inducing cytokine stimulus by compound deficiency of the tnf gene, we found that the lethality of ikappab alpha(-/-) mouse was rescued. Finally, we found that IkappaB delta provides signaling memory owing to its long half-life; it integrates the inflammatory history of the cell to dampen NF-kappaB responsiveness during sequential stimulation events.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
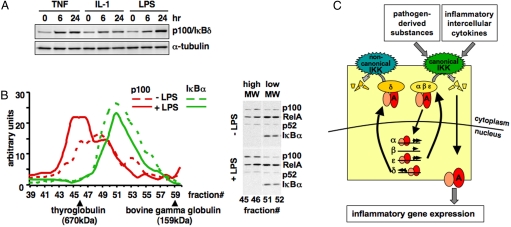
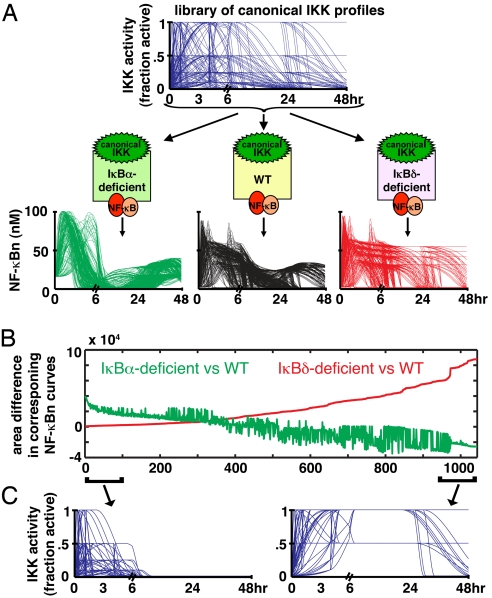
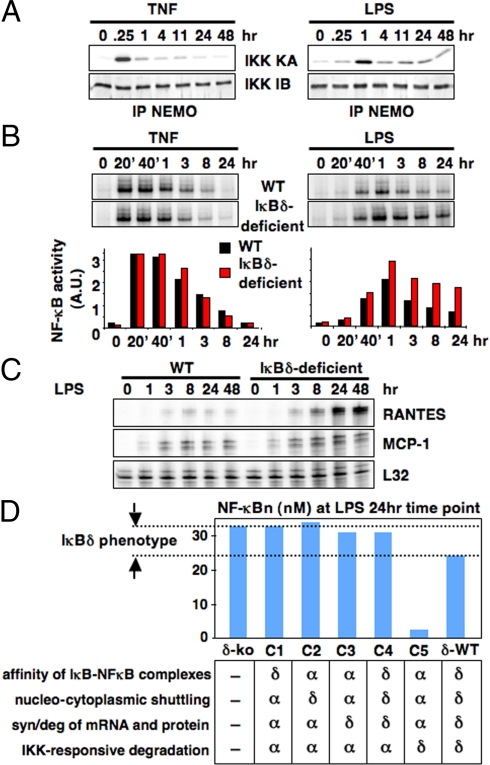
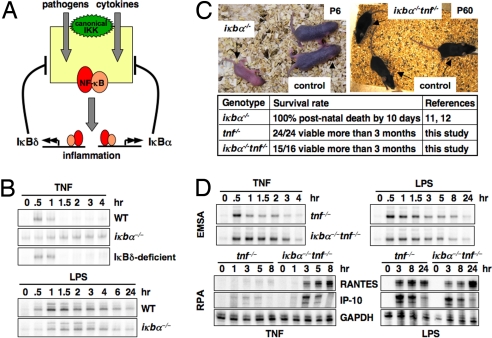
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
