

Tumultuous relationship between the human immunodeficiency virus type 1 viral infectivity factor (Vif) and the human APOBEC-3G and APOBEC-3F restriction factors
- PMID: 19487726
- PMCID: PMC2698415
- DOI: 10.1128/MMBR.00040-08
Tumultuous relationship between the human immunodeficiency virus type 1 viral infectivity factor (Vif) and the human APOBEC-3G and APOBEC-3F restriction factors
Abstract
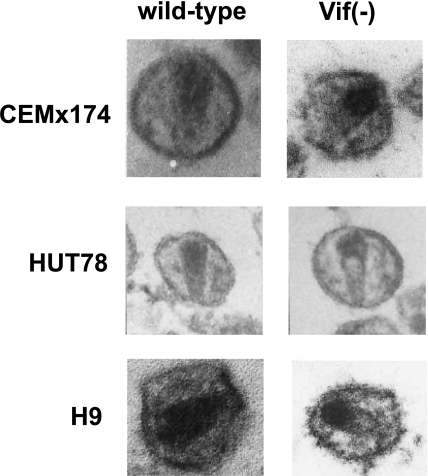
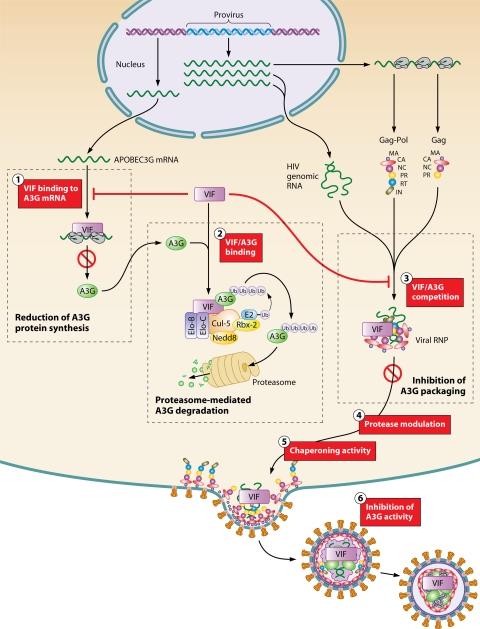
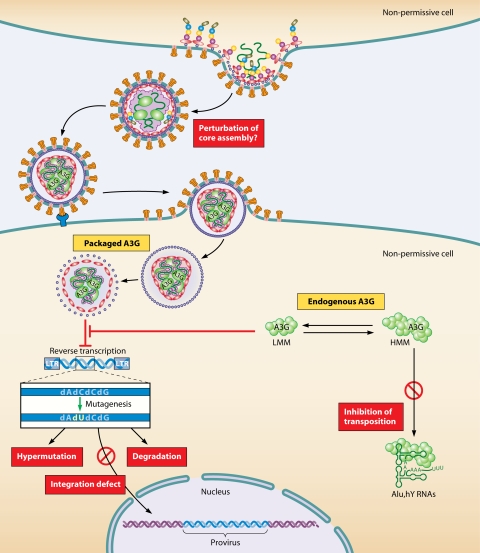
The viral infectivity factor (Vif) is dispensable for human immunodeficiency virus type 1 (HIV-1) replication in so-called permissive cells but is required for replication in nonpermissive cell lines and for pathogenesis. Virions produced in the absence of Vif have an aberrant morphology and an unstable core and are unable to complete reverse transcription. Recent studies demonstrated that human APOBEC-3G (hA3G) and APOBEC-3F (hA3F), which are selectively expressed in nonpermissive cells, possess strong anti-HIV-1 activity and are sufficient to confer a nonpermissive phenotype. Vif induces the degradation of hA3G and hA3F, suggesting that its main function is to counteract these cellular factors. Most studies focused on the hypermutation induced by the cytidine deaminase activity of hA3G and hA3F and on their Vif-induced degradation by the proteasome. However, recent studies suggested that several mechanisms are involved both in the antiviral activity of hA3G and hA3F and in the way Vif counteracts these antiviral factors. Attempts to reconcile the studies involving Vif in virus assembly and stability with these recent findings suggest that hA3G and hA3F partially exert their antiviral activity independently of their catalytic activity by destabilizing the viral core and the reverse transcription complex, possibly by interfering with the assembly and/or maturation of the viral particles. Vif could then counteract hA3G and hA3F by excluding them from the viral assembly intermediates through competition for the viral genomic RNA, by regulating the proteolytic processing of Pr55(Gag), by enhancing the efficiency of the reverse transcription process, and by inhibiting the enzymatic activities of hA3G and hA3F.
Figures
References
-
- Abudu, A., A. Takaori-Kondo, T. Izumi, K. Shirakawa, M. Kobayashi, A. Sasada, K. Fukunaga, and T. Uchiyama. 2006. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol. 161565-1570. - PubMed
-
- Aguiar, R. S., N. Lovsin, A. Tanuri, and B. M. Peterlin. 2008. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 2832518-2525. - PubMed
-
- Akari, H., M. Fujita, S. Kao, M. A. Khan, M. Shehu-Xhilaga, A. Adachi, and K. Strebel. 2004. High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site. J. Biol. Chem. 27912355-12362. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
