

Molecular disruption of RAD50 sensitizes human tumor cells to cisplatin-based chemotherapy
- PMID: 19487811
- PMCID: PMC2701852
- DOI: 10.1172/JCI33816
Molecular disruption of RAD50 sensitizes human tumor cells to cisplatin-based chemotherapy
Erratum in
- J Clin Invest. 2012 Nov;122(11):4300
Abstract
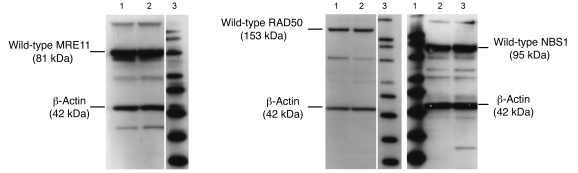
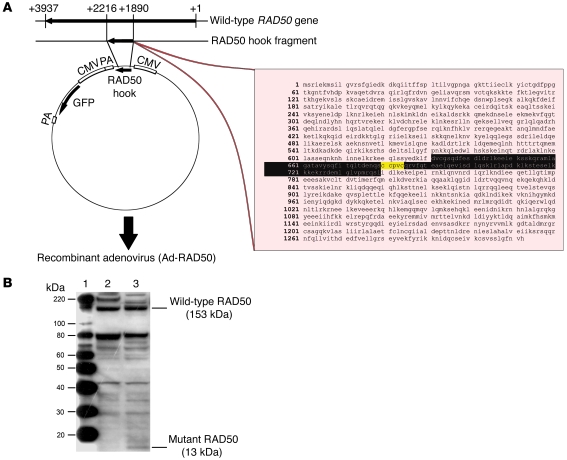
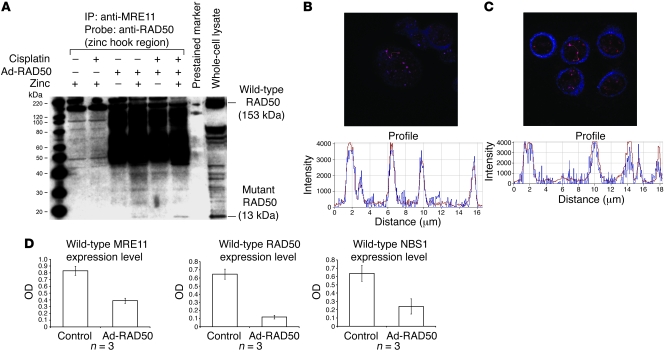
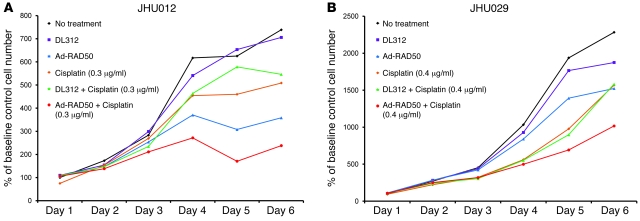
Platinum-based drugs that induce DNA damage are commonly used first-line chemotherapy agents for testicular, bladder, head and neck, lung, esophageal, stomach, and ovarian cancers. The inherent resistance of tumors to DNA damage often limits the therapeutic efficacy of these agents, such as cisplatin. An enhanced DNA repair and telomere maintenance response by the Mre11/Rad50/Nbs1 (MRN) complex is critical in driving this chemoresistance. We hypothesized therefore that the targeted impairment of native cellular MRN function could sensitize tumor cells to cisplatin. To test this, we designed what we believe to be a novel dominant-negative adenoviral vector containing a mutant RAD50 gene that significantly downregulated MRN expression and markedly disrupted MRN function in human squamous cell carcinoma cells. A combination of cisplatin and mutant RAD50 therapy produced significant tumor cytotoxicity in vitro, with a corresponding increase in DNA damage and telomere shortening. In cisplatin-resistant human squamous cell cancer xenografts in nude mice, this combination therapy caused dramatic tumor regression with increased apoptosis. Our findings suggest the use of targeted RAD50 disruption as what we believe to be a novel chemosensitizing approach for cancer therapy in the context of chemoresistance. This strategy is potentially applicable to several types of malignant tumors that demonstrate chemoresistance and may positively impact the treatment of these patients.
Figures
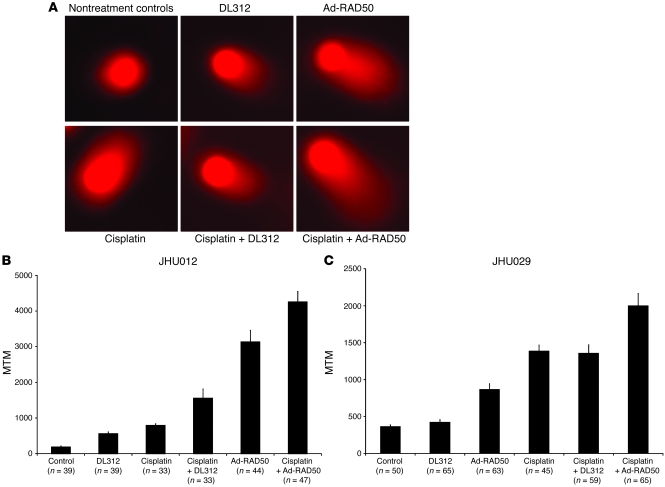
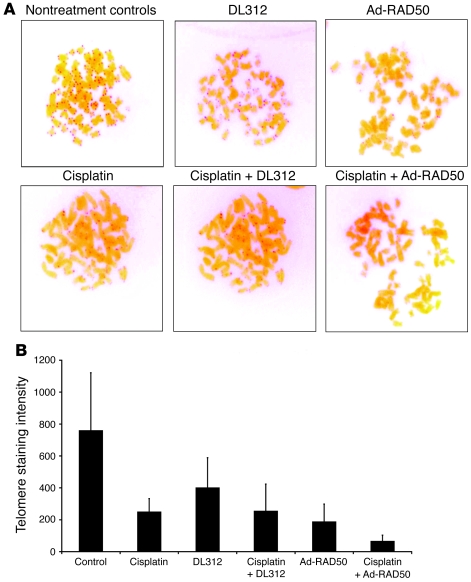
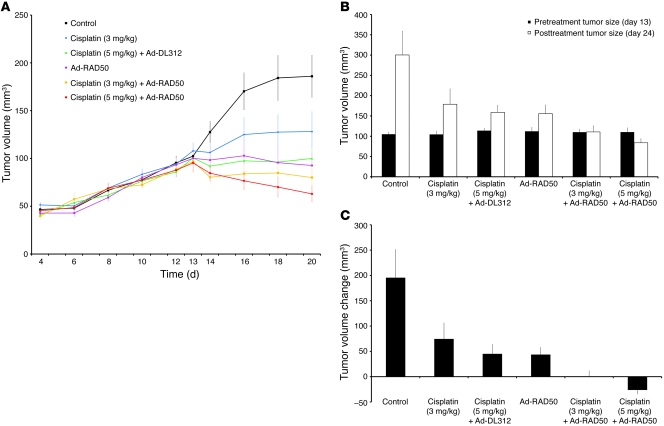
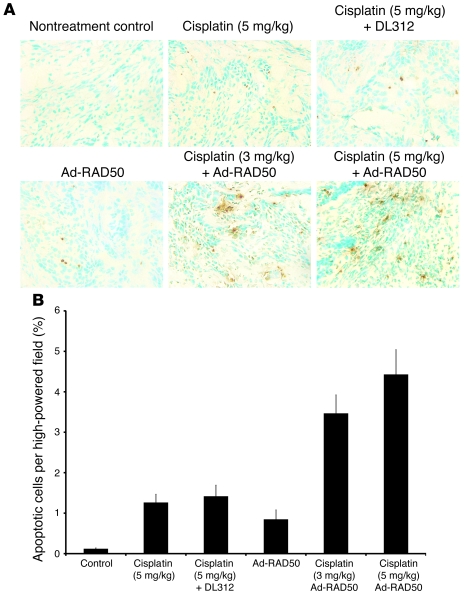
References
-
- Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J. Biol. Chem. 1994;269:787–790. - PubMed
-
- Jacobs C., et al. A phase III randomized study comparing cisplatin and fluorouracil as single agents and in combination for advanced squamous cell carcinoma of the head and neck. J. Clin. Oncol. 1992;10:257–263. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
