

Activin A is essential for neurogenesis following neurodegeneration
- PMID: 19489097
- PMCID: PMC2733378
- DOI: 10.1002/stem.80
Activin A is essential for neurogenesis following neurodegeneration
Abstract
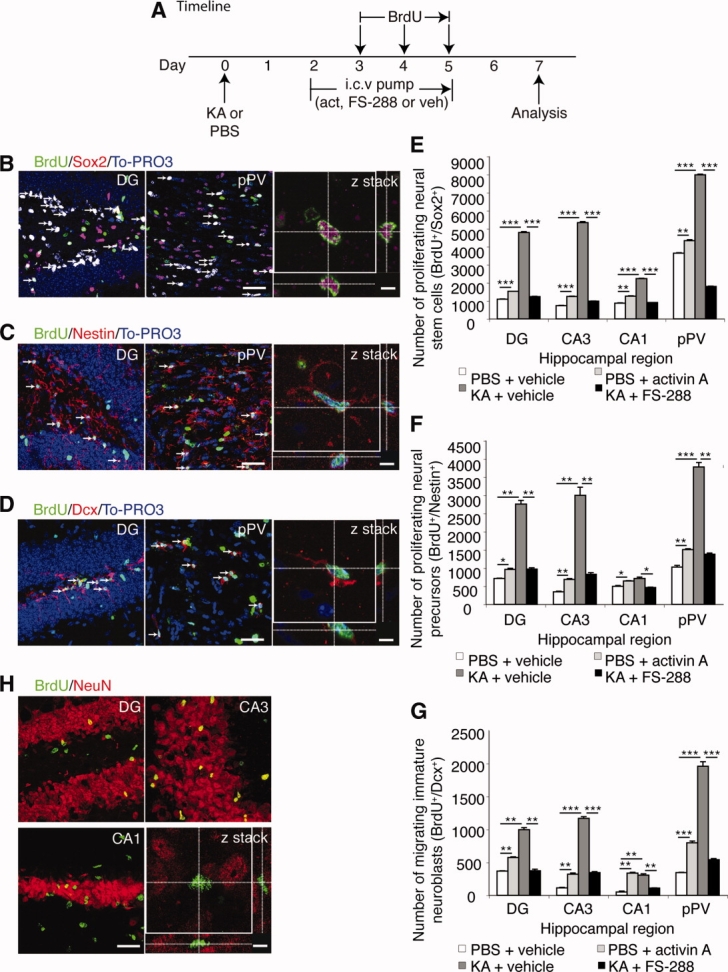
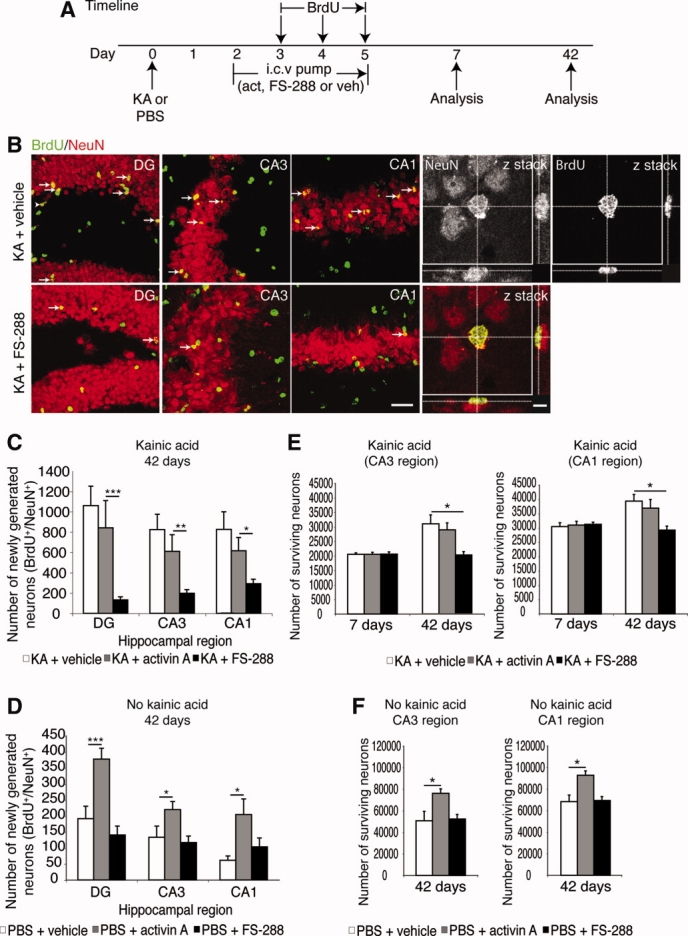
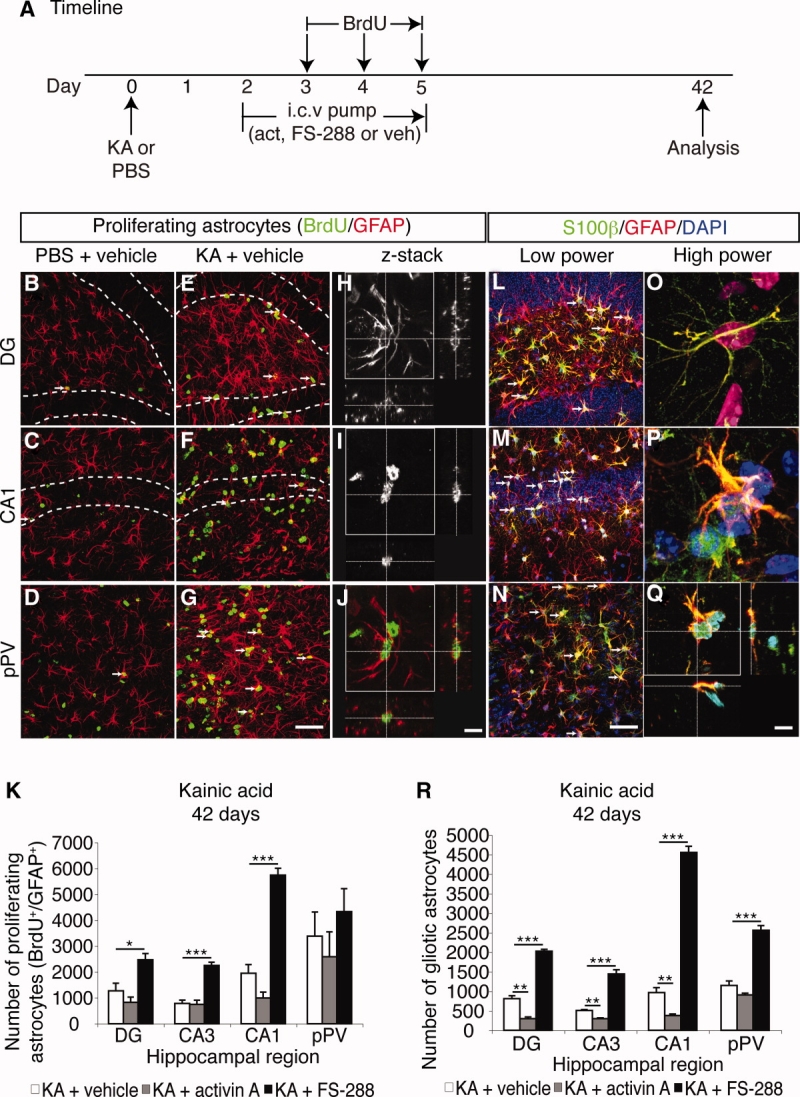
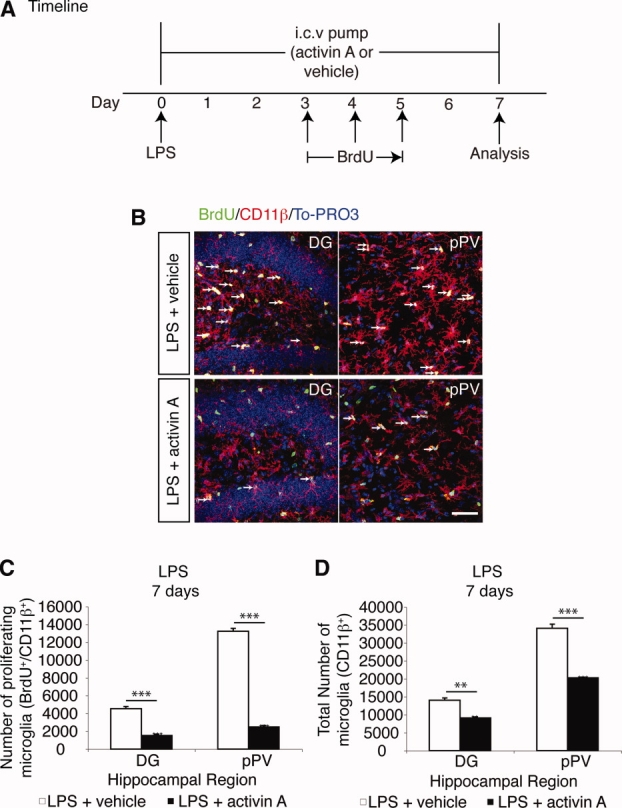
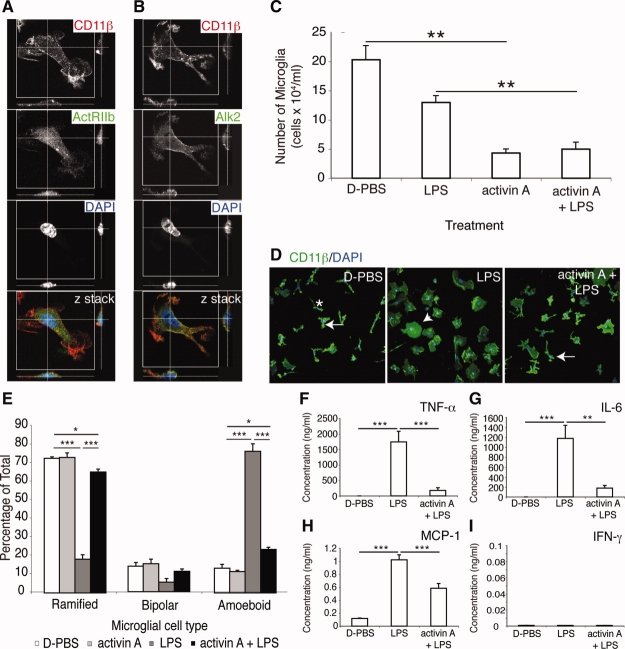
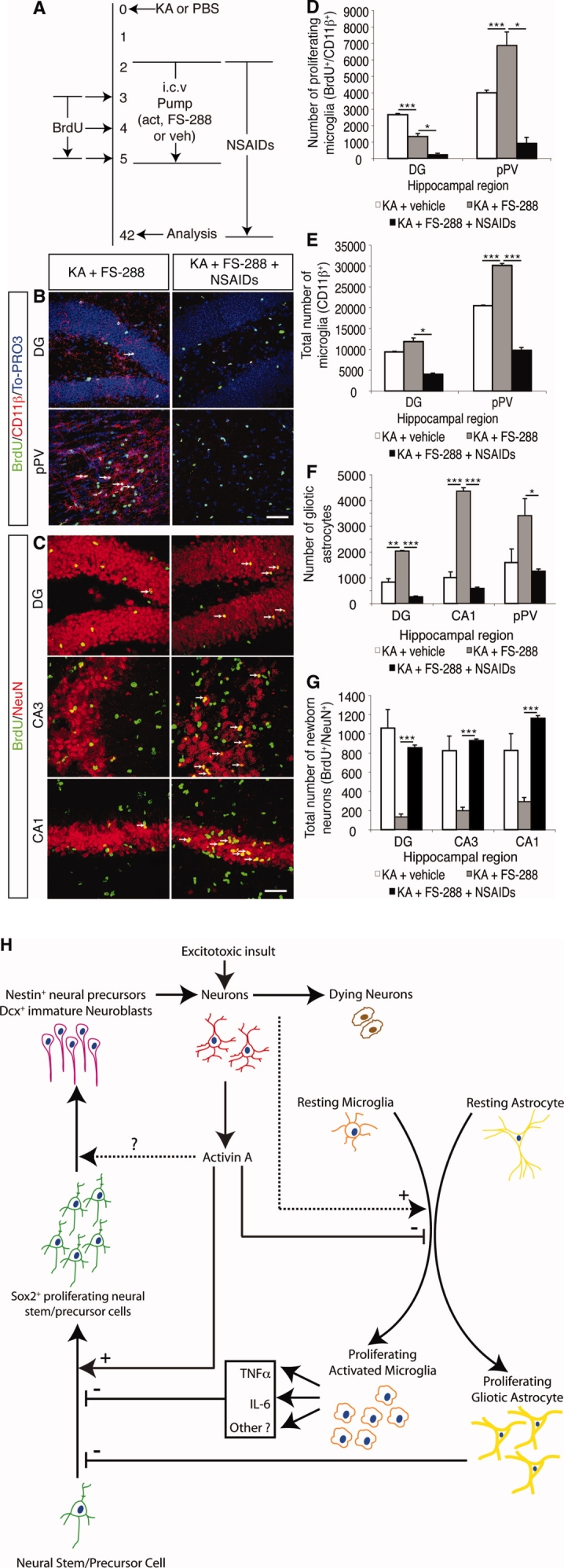
It has long been proposed that excitotoxicity contributes to nerve cell death in neurodegenerative diseases. Activin A, a member of the transforming growth factor-beta superfamily, is expressed by neurons following excitotoxicity. We show for the first time that this activin A expression is essential for neurogenesis to proceed following neurodegeneration. We found that intraventricular infusion of activin A increased the number of newborn neurons in the dentate gyrus, CA3, and CA1 layers of the normal adult hippocampus and also, following lipopolysaccharide administration, had a potent inhibitory effect on gliosis in vivo and on microglial proliferation in vivo and in vitro. Consistent with the role of activin A in regulating central nervous system inflammation and neurogenesis, intraventricular infusion of follistatin, an activin A antagonist, profoundly impaired neurogenesis and increased the number of microglia and reactive astrocytes following onset of kainic acid-induced neurodegeneration. These results show that inhibiting endogenous activin A is permissive for a potent underlying inflammatory response to neurodegeneration. We demonstrate that the anti-inflammatory actions of activin A account for its neurogenic effects following neurodegeneration because co-administration of nonsteroidal anti-inflammatory drugs reversed follistatin's inhibitory effects on neurogenesis in vivo. Our work indicates that activin A, perhaps working in conjunction with other transforming growth factor-beta superfamily molecules, is essential for neurogenesis in the adult central nervous system following excitotoxic neurodegeneration and suggests that neurons can regulate regeneration by suppressing the inflammatory response, a finding with implications for understanding and treating acute and chronic neurodegenerative diseases.
Figures

References
-
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. - PubMed
-
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. - PubMed
-
- Abdipranoto A, Wu A, Stayte S, et al. The role of neurogenesis in neurodegenerative diseases and its implications for therapeutic development. CNS Neurol Disord Drug Targets. 2008;7:187–210. - PubMed
-
- Ikonomidou C, Turski L. Excitotoxicity and neurodegenerative diseases. Curr Opin Neurol. 1995;8:487–497. - PubMed
-
- Wang Q, Yu S, Simonyi A, et al. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31:3–16. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
