

Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease
- PMID: 19491104
- PMCID: PMC2742856
- DOI: 10.1074/jbc.M109.014472
Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease
Abstract
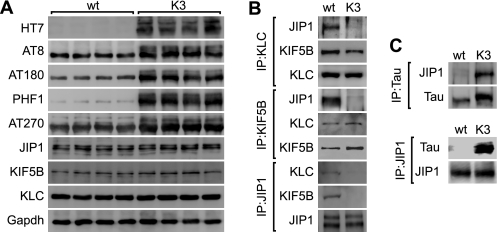
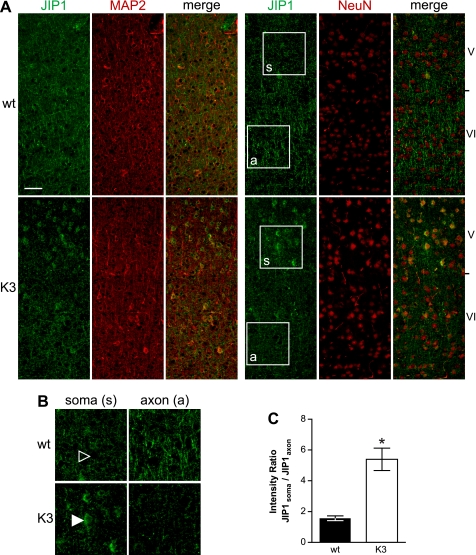
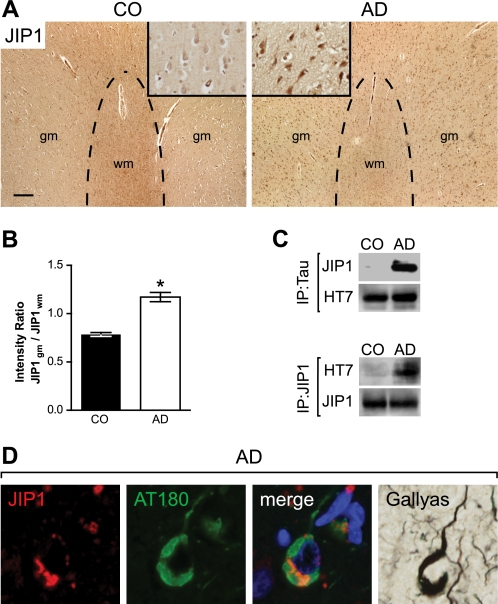
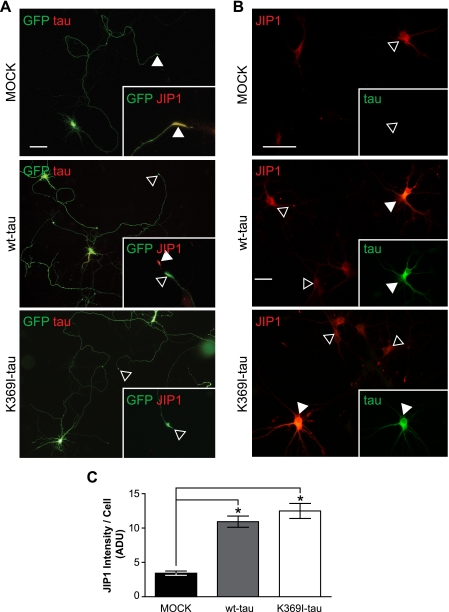
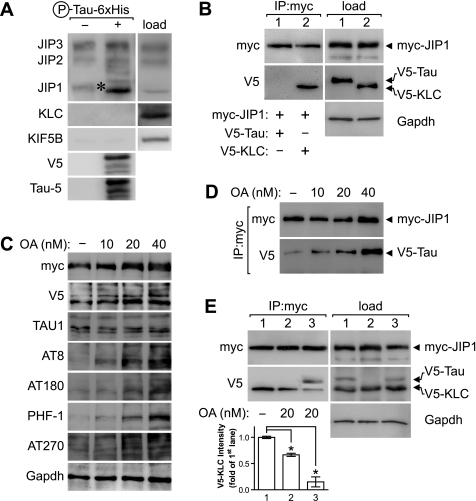
In Alzheimer disease (AD) and frontotemporal dementia the microtubule-associated protein Tau becomes progressively hyperphosphorylated, eventually forming aggregates. However, how Tau dysfunction is associated with functional impairment is only partly understood, especially at early stages when Tau is mislocalized but has not yet formed aggregates. Impaired axonal transport has been proposed as a potential pathomechanism, based on cellular Tau models and Tau transgenic mice. We recently reported K369I mutant Tau transgenic K3 mice with axonal transport defects that suggested a cargo-selective impairment of kinesin-driven anterograde transport by Tau. Here, we show that kinesin motor complex formation is disturbed in the K3 mice. We show that under pathological conditions hyperphosphorylated Tau interacts with c-Jun N-terminal kinase- interacting protein 1 (JIP1), which is associated with the kinesin motor protein complex. As a result, transport of JIP1 into the axon is impaired, causing JIP1 to accumulate in the cell body. Because we found trapping of JIP1 and a pathological Tau/JIP1 interaction also in AD brain, this may have pathomechanistic implications in diseases with a Tau pathology. This is supported by JIP1 sequestration in the cell body of Tau-transfected primary neuronal cultures. The pathological Tau/JIP1 interaction requires phosphorylation of Tau, and Tau competes with the physiological binding of JIP1 to kinesin light chain. Because JIP1 is involved in regulating cargo binding to kinesin motors, our findings may, at least in part, explain how hyperphosphorylated Tau mediates impaired axonal transport in AD and frontotemporal dementia.
Figures
References
-
- Lee V. M., Goedert M., Trojanowski J. Q. (2001) Annu. Rev. Neurosci. 24, 1121–1159 - PubMed
-
- Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989) Neuron 3, 519–526 - PubMed
-
- Alonso A. C., Grundke-Iqbal I., Iqbal K. (1996) Nat Med 2, 783–787 - PubMed
-
- Chen F., David D., Ferrari A., Götz J. (2004) Curr. Drug Targets 5, 503–515 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
