

Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication
- PMID: 19494020
- PMCID: PMC2715773
- DOI: 10.1128/JVI.02672-08
Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication
Abstract
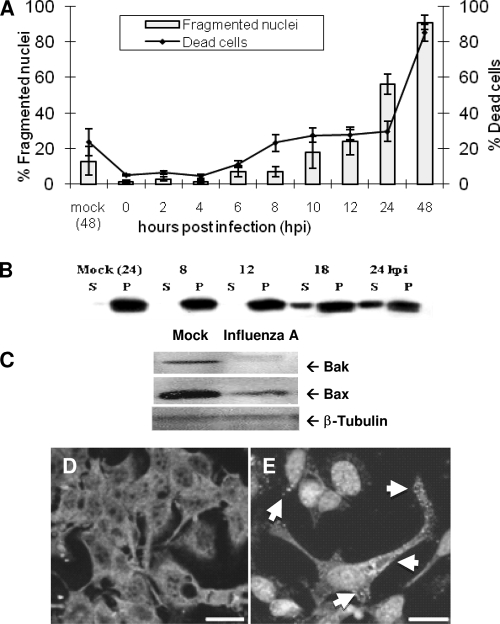
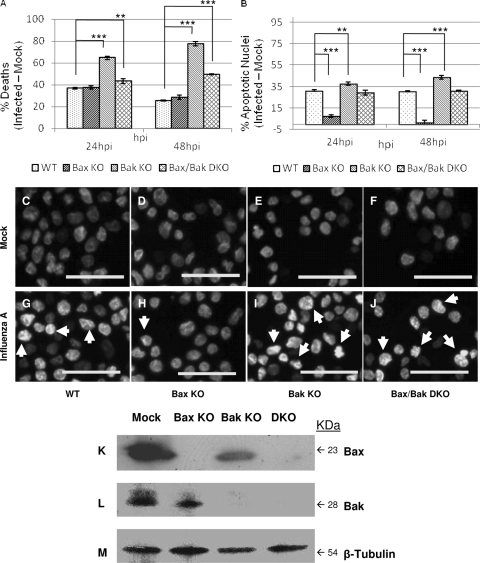
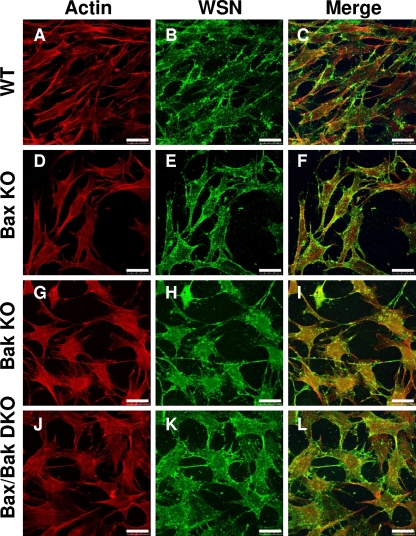
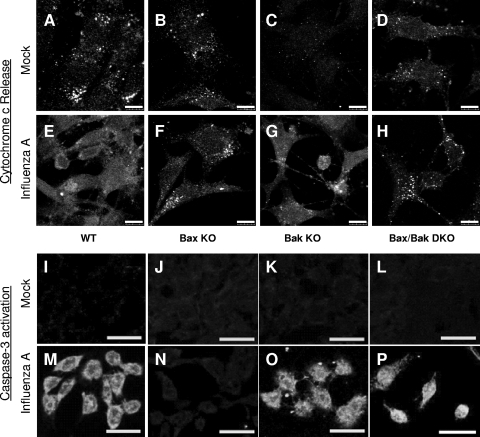
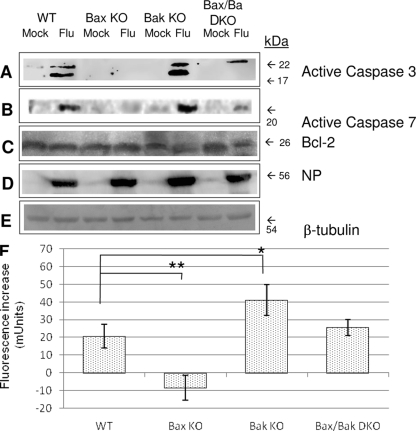
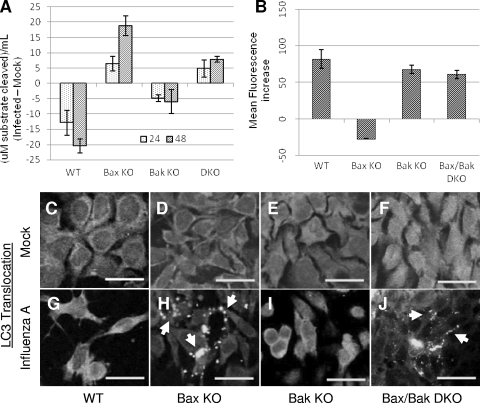
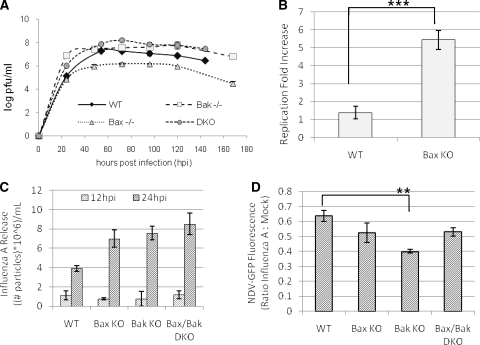
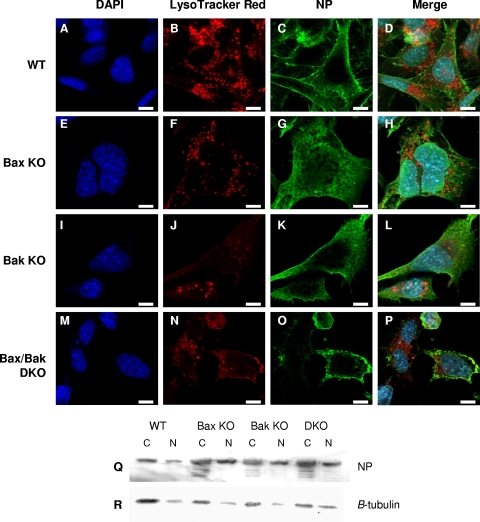
The ectopic overexpression of Bcl-2 restricts both influenza A virus-induced apoptosis and influenza A virus replication in MDCK cells, thus suggesting a role for Bcl-2 family members during infection. Here we report that influenza A virus cannot establish an apoptotic response without functional Bax, a downstream target of Bcl-2, and that both Bax and Bak are directly involved in influenza A virus replication and virus-induced cell death. Bak is substantially downregulated during influenza A virus infection in MDCK cells, and the knockout of Bak in mouse embryonic fibroblasts yields a dramatic rise in the rate of apoptotic death and a corresponding increase in levels of virus replication, suggesting that Bak suppresses both apoptosis and the replication of virus and that the virus suppresses Bak. Bax, however, is activated and translocates from the cytosol to the mitochondria; this activation is required for the efficient induction of apoptosis and virus replication. The knockout of Bax in mouse embryonic fibroblasts blocks the induction of apoptosis, restricts the infection-mediated activation of executioner caspases, and inhibits virus propagation. Bax knockout cells still die but by an alternative death pathway displaying characteristics of autophagy, similarly to our previous observation that influenza A virus infection in the presence of a pancaspase inhibitor leads to an increase in levels of autophagy. The knockout of Bax causes a retention of influenza A virus NP within the nucleus. We conclude that the cell and virus struggle to control apoptosis and autophagy, as appropriately timed apoptosis is important for the replication of influenza A virus.
Figures
Similar articles
-
Autophagy activation is required for influenza A virus-induced apoptosis and replication.Biochim Biophys Acta Mol Cell Res. 2018 Feb;1865(2):364-378. doi: 10.1016/j.bbamcr.2017.10.014. Epub 2017 Nov 3. Biochim Biophys Acta Mol Cell Res. 2018. PMID: 29108912
-
Bax and Bak are required for apoptosis induction by sulforaphane, a cruciferous vegetable-derived cancer chemopreventive agent.Cancer Res. 2005 Mar 1;65(5):2035-43. doi: 10.1158/0008-5472.CAN-04-3616. Cancer Res. 2005. PMID: 15753404
-
Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication.Cell Death Dis. 2015 Dec 17;6(12):e2018. doi: 10.1038/cddis.2015.360. Cell Death Dis. 2015. PMID: 26673663 Free PMC article.
-
Influenza A virus-induced apoptosis and virus propagation.Apoptosis. 2020 Feb;25(1-2):1-11. doi: 10.1007/s10495-019-01575-3. Apoptosis. 2020. PMID: 31667646 Review.
-
BCL-2 proteins and apoptosis: Recent insights and unknowns.Biochem Biophys Res Commun. 2018 May 27;500(1):26-34. doi: 10.1016/j.bbrc.2017.06.190. Epub 2017 Jul 1. Biochem Biophys Res Commun. 2018. PMID: 28676391 Review.
Cited by
-
The Wheel of p53 Helps to Drive the Immune System.Int J Mol Sci. 2023 Apr 21;24(8):7645. doi: 10.3390/ijms24087645. Int J Mol Sci. 2023. PMID: 37108808 Free PMC article. Review.
-
Bik Mediates Caspase-Dependent Cleavage of Viral Proteins to Promote Influenza A Virus Infection.Am J Respir Cell Mol Biol. 2016 May;54(5):664-73. doi: 10.1165/rcmb.2015-0133OC. Am J Respir Cell Mol Biol. 2016. PMID: 26437021 Free PMC article.
-
Stress Granule-Inducing Eukaryotic Translation Initiation Factor 4A Inhibitors Block Influenza A Virus Replication.Viruses. 2017 Dec 18;9(12):388. doi: 10.3390/v9120388. Viruses. 2017. PMID: 29258238 Free PMC article.
-
Anti-Influenza Activity of the Ribonuclease Binase: Cellular Targets Detected by Quantitative Proteomics.Int J Mol Sci. 2020 Nov 5;21(21):8294. doi: 10.3390/ijms21218294. Int J Mol Sci. 2020. PMID: 33167434 Free PMC article.
-
The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections.Virulence. 2019 Dec;10(1):376-413. doi: 10.1080/21505594.2019.1605803. Virulence. 2019. PMID: 30966844 Free PMC article. Review.
References
-
- Brydon, E. W. A., S. J. Morris, and C. Sweet. 2005. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 29837-850. - PubMed
-
- Brydon, E. W. A., H. Smith, and C. Sweet. 2003. Influenza A virus-induced apoptosis in bronchiolar epithelial (NCI-H292) cells limits pro-inflammatory cytokine release. Soc. Gen. Microbiol. 842389-2400. - PubMed
-
- Chen, W., P. A. Calvo, and D. Malide. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 71306-1312. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
