

Golgi function and dysfunction in the first COG4-deficient CDG type II patient
- PMID: 19494034
- PMCID: PMC2722986
- DOI: 10.1093/hmg/ddp262
Golgi function and dysfunction in the first COG4-deficient CDG type II patient
Abstract
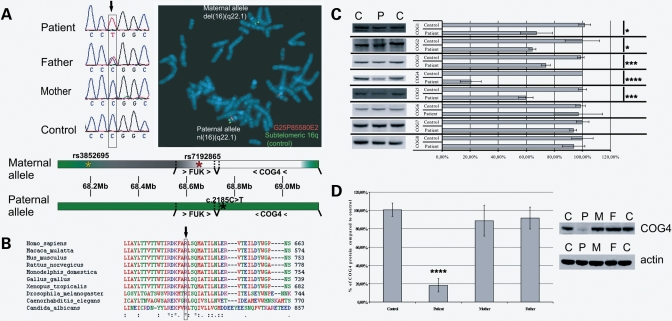
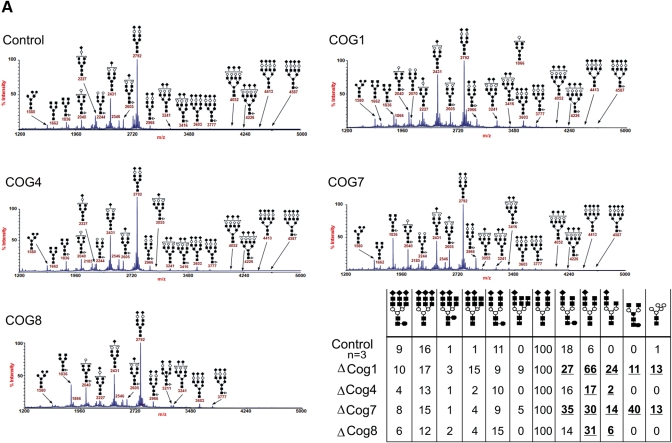
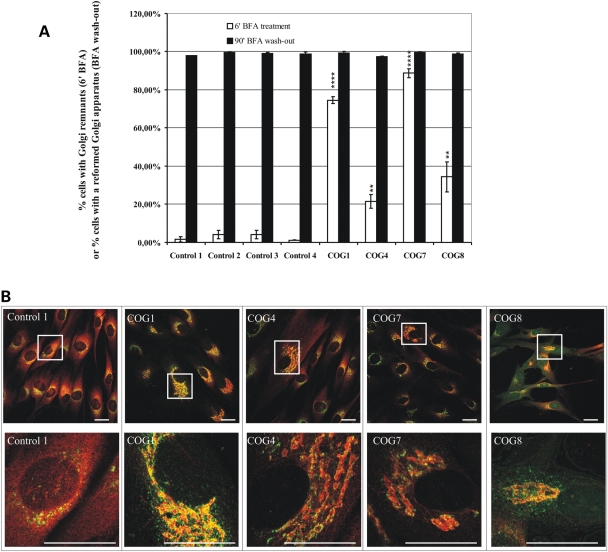
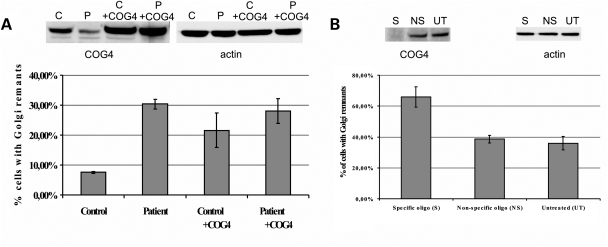
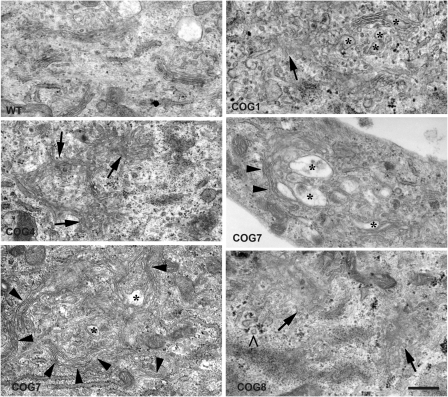
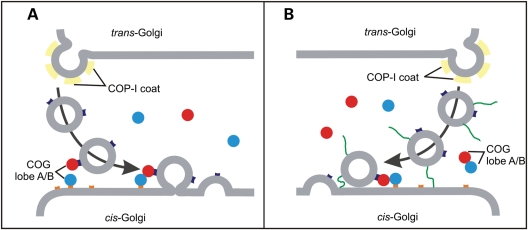
The conserved oligomeric Golgi (COG) complex is a hetero-octameric complex essential for normal glycosylation and intra-Golgi transport. An increasing number of congenital disorder of glycosylation type II (CDG-II) mutations are found in COG subunits indicating its importance in glycosylation. We report a new CDG-II patient harbouring a p.R729W missense mutation in COG4 combined with a submicroscopical deletion. The resulting downregulation of COG4 expression additionally affects expression or stability of other lobe A subunits. Despite this, full complex formation was maintained albeit to a lower extent as shown by glycerol gradient centrifugation. Moreover, our data indicate that subunits are present in a cytosolic pool and full complex formation assists tethering preceding membrane fusion. By extending this study to four other known COG-deficient patients, we now present the first comparative analysis on defects in transport, glycosylation and Golgi ultrastructure in these patients. The observed structural and biochemical abnormalities correlate with the severity of the mutation, with the COG4 mutant being the mildest. All together our results indicate that intact COG complexes are required to maintain Golgi dynamics and its associated functions. According to the current CDG nomenclature, this newly identified deficiency is designated CDG-IIj.
Figures

Similar articles
-
Deficiency of the Cog8 subunit in normal and CDG-derived cells impairs the assembly of the COG and Golgi SNARE complexes.Traffic. 2013 Oct;14(10):1065-77. doi: 10.1111/tra.12093. Epub 2013 Jul 31. Traffic. 2013. PMID: 23865579 Free PMC article.
-
Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II.Proc Natl Acad Sci U S A. 2006 Mar 7;103(10):3764-9. doi: 10.1073/pnas.0507685103. Epub 2006 Feb 28. Proc Natl Acad Sci U S A. 2006. PMID: 16537452 Free PMC article.
-
Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder.Nat Med. 2004 May;10(5):518-23. doi: 10.1038/nm1041. Epub 2004 Apr 25. Nat Med. 2004. PMID: 15107842
-
Golgi inCOGnito: From vesicle tethering to human disease.Biochim Biophys Acta Gen Subj. 2020 Nov;1864(11):129694. doi: 10.1016/j.bbagen.2020.129694. Epub 2020 Jul 27. Biochim Biophys Acta Gen Subj. 2020. PMID: 32730773 Free PMC article. Review.
-
Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation.Mol Genet Metab. 2008 Jan;93(1):15-21. doi: 10.1016/j.ymgme.2007.08.118. Epub 2007 Sep 29. Mol Genet Metab. 2008. PMID: 17904886 Review.
Cited by
-
Fetal bovine serum impacts the observed N-glycosylation defects in TMEM165 KO HEK cells.J Inherit Metab Dis. 2020 Mar;43(2):357-366. doi: 10.1002/jimd.12161. Epub 2019 Oct 1. J Inherit Metab Dis. 2020. PMID: 31415112 Free PMC article.
-
Modeling Congenital Disorders of N-Linked Glycoprotein Glycosylation in Drosophila melanogaster.Front Genet. 2018 Oct 2;9:436. doi: 10.3389/fgene.2018.00436. eCollection 2018. Front Genet. 2018. PMID: 30333856 Free PMC article. Review.
-
Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation.Front Genet. 2020 Feb 27;11:168. doi: 10.3389/fgene.2020.00168. eCollection 2020. Front Genet. 2020. PMID: 32174980 Free PMC article.
-
Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function.Biochem Biophys Res Commun. 2014 Oct 17;453(2):243-53. doi: 10.1016/j.bbrc.2014.06.067. Epub 2014 Jun 24. Biochem Biophys Res Commun. 2014. PMID: 24971539 Free PMC article. Review.
-
Deficiency of the Cog8 subunit in normal and CDG-derived cells impairs the assembly of the COG and Golgi SNARE complexes.Traffic. 2013 Oct;14(10):1065-77. doi: 10.1111/tra.12093. Epub 2013 Jul 31. Traffic. 2013. PMID: 23865579 Free PMC article.
References
-
- Lippincott-Schwartz J., Zaal K.J. Cell cycle maintenance and biogenesis of the Golgi complex. Histochem. Cell Biol. 2000;114:93–103. - PubMed
-
- Yan A., Lennarz W.J. Unraveling the mechanism of protein N-glycosylation. J. Biol. Chem. 280:3121–3124. - PubMed
-
- Holthuis J.C., Pomorski T., Raggers R.J., Sprong H., Van Meer G. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 2001;81:1689–1723. - PubMed
-
- Dunphy W.G., Rothman J.E. Compartmental organization of the Golgi stack. Cell. 1985;42:13–21. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
