

Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53
- PMID: 19494119
- PMCID: PMC2740446
- DOI: 10.1074/jbc.M109.026096
Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53
Abstract
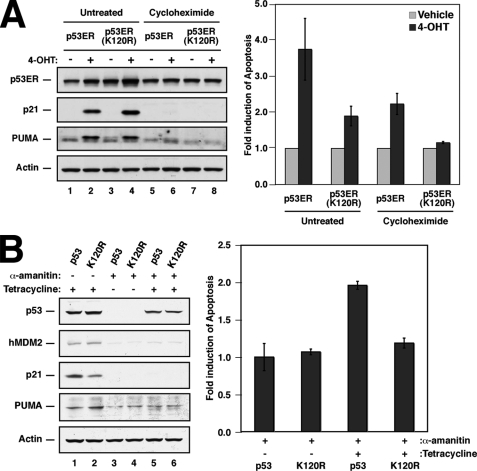
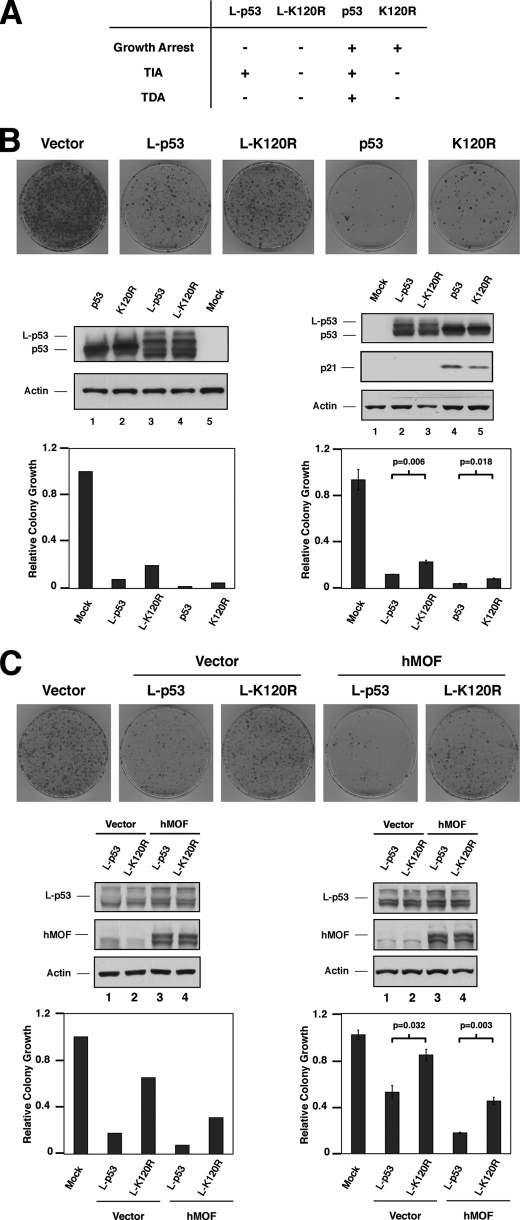
The tumor suppressor p53 induces apoptosis by altering the transcription of pro-apoptotic targets in the nucleus and by a direct, nontranscriptional role at the mitochondria. Although the post-translational modifications regulating nuclear apoptotic functions of p53 have been thoroughly characterized, little is known of how transcription-independent functions are controlled. We and others identified acetylation of the p53 DNA binding domain at lysine 120 as a critical event in apoptosis induction. Although initial studies showed that Lys-120 acetylation plays a role in p53 function in the nucleus, we report here a role for Lys-120 acetylation in transcription-independent apoptosis. We demonstrate that the Lys-120-acetylated isoform of p53 is enriched at mitochondria. The acetylation of Lys-120 does not appear to regulate the ability of p53 to interact with the pro-apoptotic proteins BCL-XL and BAK. However, displacement of the inhibitory MCL-1 protein from BAK is compromised when Lys-120 acetylation is blocked. Functional studies show that mutation of Lys-120 to a nonacetylated residue, as occurs in human cancer, inhibits transcription-independent apoptosis, and enforced acetylation of Lys-120 enhances transcription-independent apoptosis. These data support a model whereby Lys-120 acetylation contributes to both the transcription-dependent and -independent apoptotic pathways induced by p53.
Figures
Similar articles
-
p14(ARF)-induced apoptosis in p53 protein-deficient cells is mediated by BH3-only protein-independent derepression of Bak protein through down-regulation of Mcl-1 and Bcl-xL proteins.J Biol Chem. 2012 May 18;287(21):17343-17352. doi: 10.1074/jbc.M111.314898. Epub 2012 Feb 21. J Biol Chem. 2012. PMID: 22354970 Free PMC article.
-
Bax/Bak activation in the absence of Bid, Bim, Puma, and p53.Cell Death Dis. 2016 Jun 16;7(6):e2266. doi: 10.1038/cddis.2016.167. Cell Death Dis. 2016. PMID: 27310874 Free PMC article.
-
miR-128 exerts pro-apoptotic effect in a p53 transcription-dependent and -independent manner via PUMA-Bak axis.Cell Death Dis. 2013 Mar 14;4(3):e542. doi: 10.1038/cddis.2013.46. Cell Death Dis. 2013. PMID: 23492773 Free PMC article.
-
The ARF/oncogene pathway activates p53 acetylation within the DNA binding domain.Cell Cycle. 2007 Jun 1;6(11):1304-6. doi: 10.4161/cc.6.11.4343. Epub 2007 Jun 24. Cell Cycle. 2007. PMID: 17534149 Review.
-
Structural insights into the transcription-independent apoptotic pathway of p53.BMB Rep. 2014 Mar;47(3):167-72. doi: 10.5483/bmbrep.2014.47.3.261. BMB Rep. 2014. PMID: 24499665 Free PMC article. Review.
Cited by
-
A Different View for an Old Disease: NEDDylation and Other Ubiquitin-Like Post-Translational Modifications in Chronic Lymphocytic Leukemia.Front Oncol. 2021 Sep 23;11:729550. doi: 10.3389/fonc.2021.729550. eCollection 2021. Front Oncol. 2021. PMID: 34631557 Free PMC article. Review.
-
Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis.Cell Death Differ. 2018 Jan;25(1):190-203. doi: 10.1038/cdd.2017.143. Epub 2017 Sep 22. Cell Death Differ. 2018. PMID: 28937686 Free PMC article.
-
Nutlin's two roads toward apoptosis.Cancer Biol Ther. 2010 Sep 15;10(6):579-81. doi: 10.4161/cbt.10.6.13127. Epub 2010 Sep 24. Cancer Biol Ther. 2010. PMID: 20716967 Free PMC article. No abstract available.
-
HOPS and p53: thick as thieves in life and death.Cell Cycle. 2020 Nov;19(22):2996-3003. doi: 10.1080/15384101.2020.1838772. Epub 2020 Oct 28. Cell Cycle. 2020. PMID: 33112208 Free PMC article. Review.
-
Preferred drifting along the DNA major groove and cooperative anchoring of the p53 core domain: mechanisms and scenarios.J Mol Recognit. 2010 Mar-Apr;23(2):232-40. doi: 10.1002/jmr.990. J Mol Recognit. 2010. PMID: 19856322 Free PMC article.
References
-
- Vousden K. H., Lu X. (2002) Nat. Rev. Cancer 2, 594–604 - PubMed
-
- el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) Cell 75, 817–825 - PubMed
-
- Chipuk J. E., Green D. R. (2006) Cell Death Differ. 13, 994–1002 - PubMed
-
- Kuribayashi K., El-Deiry W. S. (2008) Adv. Exp. Med. Biol. 615, 201–221 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
