

ABACAS: algorithm-based automatic contiguation of assembled sequences
- PMID: 19497936
- PMCID: PMC2712343
- DOI: 10.1093/bioinformatics/btp347
ABACAS: algorithm-based automatic contiguation of assembled sequences
Abstract
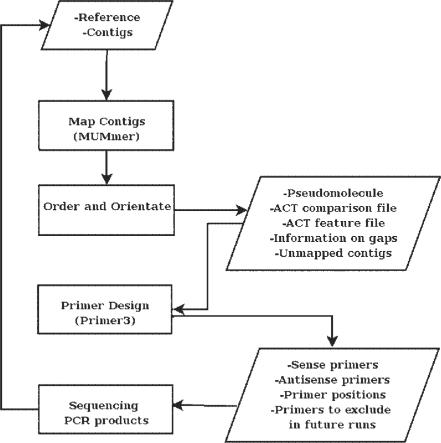
Summary: Due to the availability of new sequencing technologies, we are now increasingly interested in sequencing closely related strains of existing finished genomes. Recently a number of de novo and mapping-based assemblers have been developed to produce high quality draft genomes from new sequencing technology reads. New tools are necessary to take contigs from a draft assembly through to a fully contiguated genome sequence. ABACAS is intended as a tool to rapidly contiguate (align, order, orientate), visualize and design primers to close gaps on shotgun assembled contigs based on a reference sequence. The input to ABACAS is a set of contigs which will be aligned to the reference genome, ordered and orientated, visualized in the ACT comparative browser, and optimal primer sequences are automatically generated.
Availability and implementation: ABACAS is implemented in Perl and is freely available for download from http://abacas.sourceforge.net.
Figures
References
-
- Altschul SF, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Bartels D, et al. BACCardI–a tool for the validation of genomic assemblies, assisting genome finishing and intergenome comparison. Bioinformatics. 2005;21:853–859. - PubMed
-
- Chaisson, et al. Fragment assembly with short reads. Bioinformatics. 2004;20:2067–2074. - PubMed
-
- Gordon D, et al. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. - PubMed
