

International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family
- PMID: 19498085
- PMCID: PMC2745437
- DOI: 10.1124/pr.109.001578
International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family
Abstract
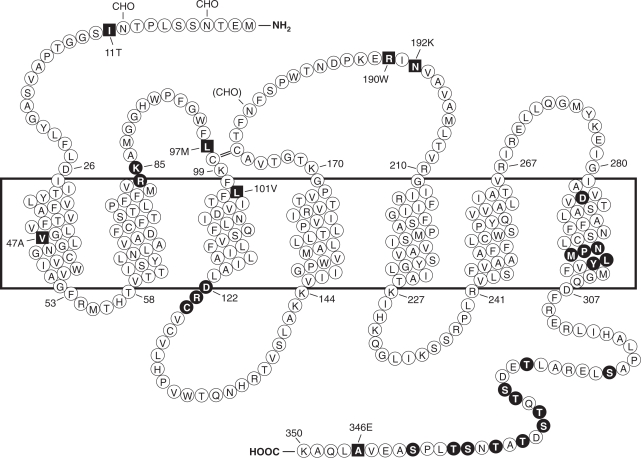
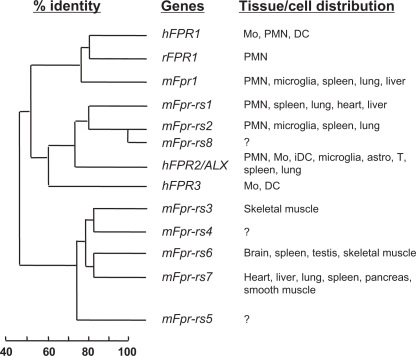
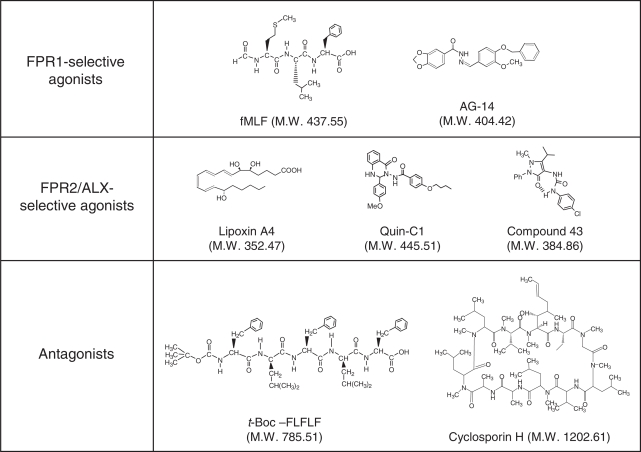
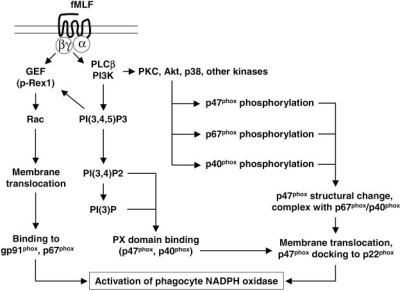
Formyl peptide receptors (FPRs) are a small group of seven-transmembrane domain, G protein-coupled receptors that are expressed mainly by mammalian phagocytic leukocytes and are known to be important in host defense and inflammation. The three human FPRs (FPR1, FPR2/ALX, and FPR3) share significant sequence homology and are encoded by clustered genes. Collectively, these receptors bind an extraordinarily numerous and structurally diverse group of agonistic ligands, including N-formyl and nonformyl peptides of different composition, that chemoattract and activate phagocytes. N-formyl peptides, which are encoded in nature only by bacterial and mitochondrial genes and result from obligatory initiation of bacterial and mitochondrial protein synthesis with N-formylmethionine, is the only ligand class common to all three human receptors. Surprisingly, the endogenous anti-inflammatory peptide annexin 1 and its N-terminal fragments also bind human FPR1 and FPR2/ALX, and the anti-inflammatory eicosanoid lipoxin A4 is an agonist at FPR2/ALX. In comparison, fewer agonists have been identified for FPR3, the third member in this receptor family. Structural and functional studies of the FPRs have produced important information for understanding the general pharmacological principles governing all leukocyte chemoattractant receptors. This article aims to provide an overview of the discovery and pharmacological characterization of FPRs, to introduce an International Union of Basic and Clinical Pharmacology (IUPHAR)-recommended nomenclature, and to discuss unmet challenges, including the mechanisms used by these receptors to bind diverse ligands and mediate different biological functions.
Figures


References
-
- Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature. 1991;353:668–670. - PubMed
-
- Agarwal S, Reynolds MA, Duckett LD, Suzuki JB. Altered free cytosolic calcium changes and neutrophil chemotaxis in patients with juvenile periodontitis. J Periodontal Res. 1989;24:149–154. - PubMed
-
- Ali H, Richardson RM, Haribabu B, Snyderman R. Chemoattractant receptor cross-desensitization. J Biol Chem. 1999;274:6027–6030. - PubMed
-
- Ali H, Richardson RM, Tomhave ED, Didsbury JR, Snyderman R. Differences in phosphorylation of formylpeptide and C5a chemoattractant receptors correlate with differences in desensitization. J Biol Chem. 1993;268:24247–24254. - PubMed
-
- Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat Immunol. 2002a;3:76–82. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
