

Role of glycogen synthase kinase-3beta in cardioprotection
- PMID: 19498210
- PMCID: PMC2726042
- DOI: 10.1161/CIRCRESAHA.109.197996
Role of glycogen synthase kinase-3beta in cardioprotection
Abstract
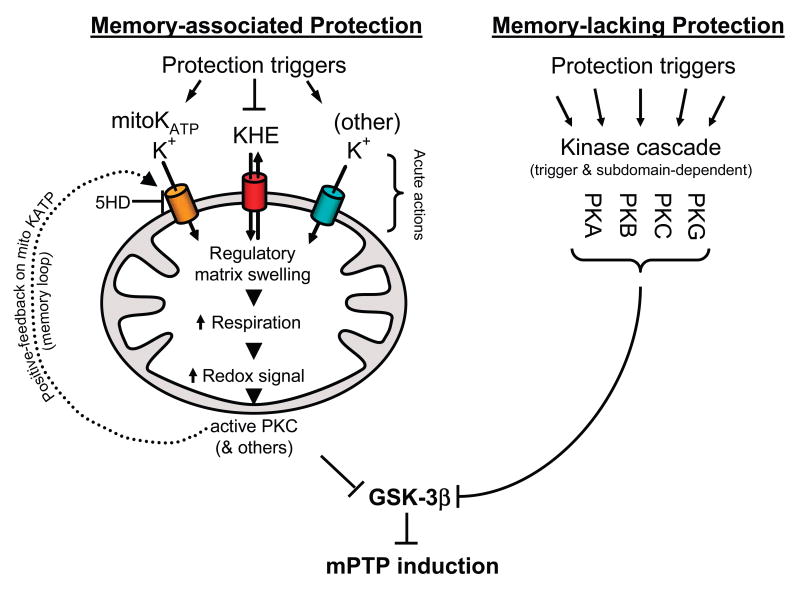
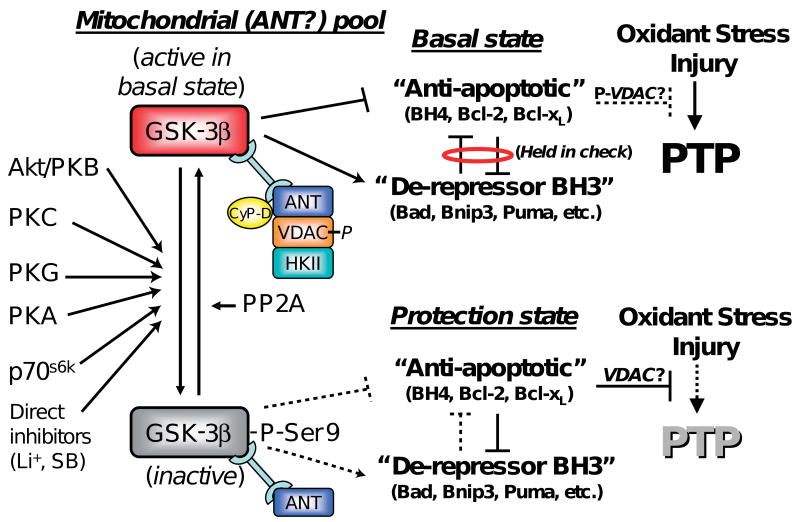
Limitation of infarct size by ischemic/pharmacological pre- and postconditioning involves activation of a complex set of cell-signaling pathways. Multiple lines of evidence implicate the mitochondrial permeability transition pore (mPTP) as a key end effector of ischemic/pharmacological pre- and postconditioning. Increasing the ROS threshold for mPTP induction enhances the resistance of cardiomyocytes to oxidant stress and results in infarct size reduction. Here, we survey and synthesize the present knowledge about the role of glycogen synthase kinase (GSK)-3beta in cardioprotection, including pre- and postconditioning. Activation of a wide spectrum of cardioprotective signaling pathways is associated with phosphorylation and inhibition of a discrete pool of GSK-3beta relevant to mitochondrial signaling. Therefore, GSK-3beta has emerged as the integration point of many of these pathways and plays a central role in transferring protective signals downstream to target(s) that act at or in proximity to the mPTP. Bcl-2 family proteins and mPTP-regulatory elements, such as adenine nucleotide translocator and cyclophilin D (possibly voltage-dependent anion channel), may be the functional downstream target(s) of GSK-3beta. Gaining a better understanding of these interactions to control and prevent mPTP induction when appropriate will enable us to decrease the negative impact of the reperfusion-induced ROS burst on the fate of mitochondria and perhaps allow us to limit propagation of damage throughout and between cells and consequently, to better limit infarct size.
Conflict of interest statement
Figures
References
-
- Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch Pathol. 1960;70:68–78. - PubMed
-
- Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–190. - PubMed
-
- Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc Res. 2006;70:191–199. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
