

A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease
- PMID: 19506092
- PMCID: PMC2752120
- DOI: 10.1101/gr.094987.109
A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease
Abstract
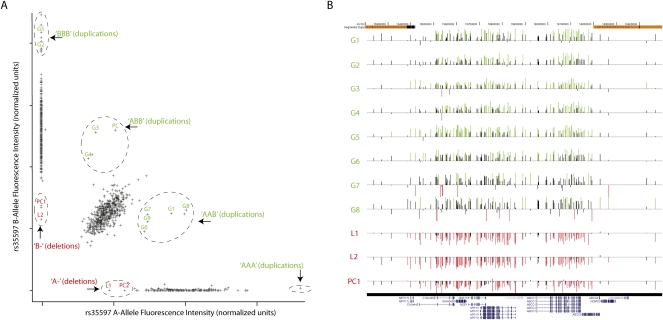
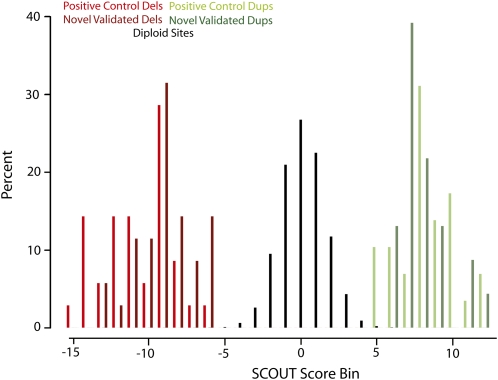
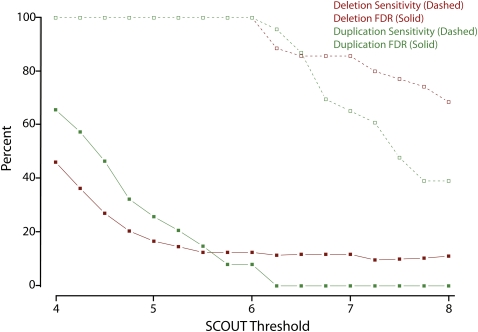
Copy-number variants (CNVs) are substantial contributors to human disease. A central challenge in CNV-disease association studies is to characterize the pathogenicity of rare and possibly incompletely penetrant events, which requires the accurate detection of rare CNVs in large numbers of individuals. Cost and throughput issues limit our ability to perform these studies. We have adapted the Illumina BeadXpress SNP genotyping assay and developed an algorithm, SNP-Conditional OUTlier detection (SCOUT), to rapidly and accurately detect both rare and common CNVs in large cohorts. This approach is customizable, cost effective, highly parallelized, and largely automated. We applied this method to screen 69 loci in 1105 children with unexplained intellectual disability, identifying pathogenic variants in 3.1% of these individuals and potentially pathogenic variants in an additional 2.3%. We identified seven individuals (0.7%) with a deletion of 16p11.2, which has been previously associated with autism. Our results widen the phenotypic spectrum of these deletions to include intellectual disability without autism. We also detected 1.65-3.4 Mbp duplications at 16p13.11 in 1.1% of affected individuals and 350 kbp deletions at 15q11.2, near the Prader-Willi/Angelman syndrome critical region, in 0.8% of affected individuals. Compared to published CNVs in controls they are significantly (P = 4.7 x 10(-5) and 0.003, respectively) enriched in these children, supporting previously published hypotheses that they are neurocognitive disease risk factors. More generally, this approach offers a previously unavailable balance between customization, cost, and throughput for analysis of CNVs and should prove valuable for targeted CNV detection in both research and diagnostic settings.
Figures
References
-
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. - PubMed
-
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Perez Jurado LA, Delle Chiaie B, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009;52:77–87. - PubMed
-
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. - PMC - PubMed
-
- Carlson CS, Smith JD, Stanaway IB, Rieder MJ, Nickerson DA. Direct detection of null alleles in SNP genotyping data. Hum Mol Genet. 2006;15:1931–1937. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous