

How eukaryotic genes are transcribed
- PMID: 19514890
- PMCID: PMC2718758
- DOI: 10.1080/10409230902858785
How eukaryotic genes are transcribed
Abstract
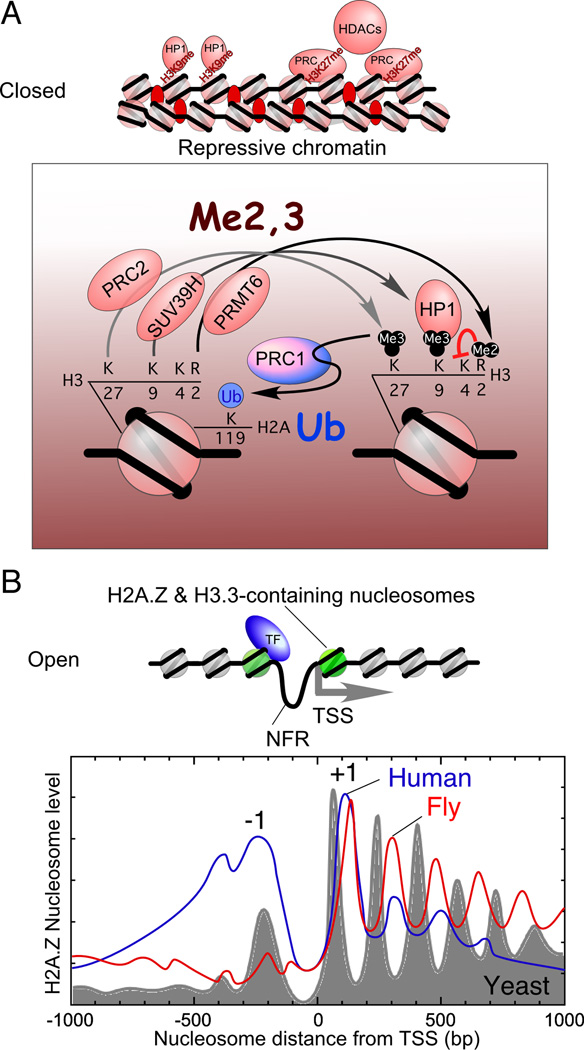
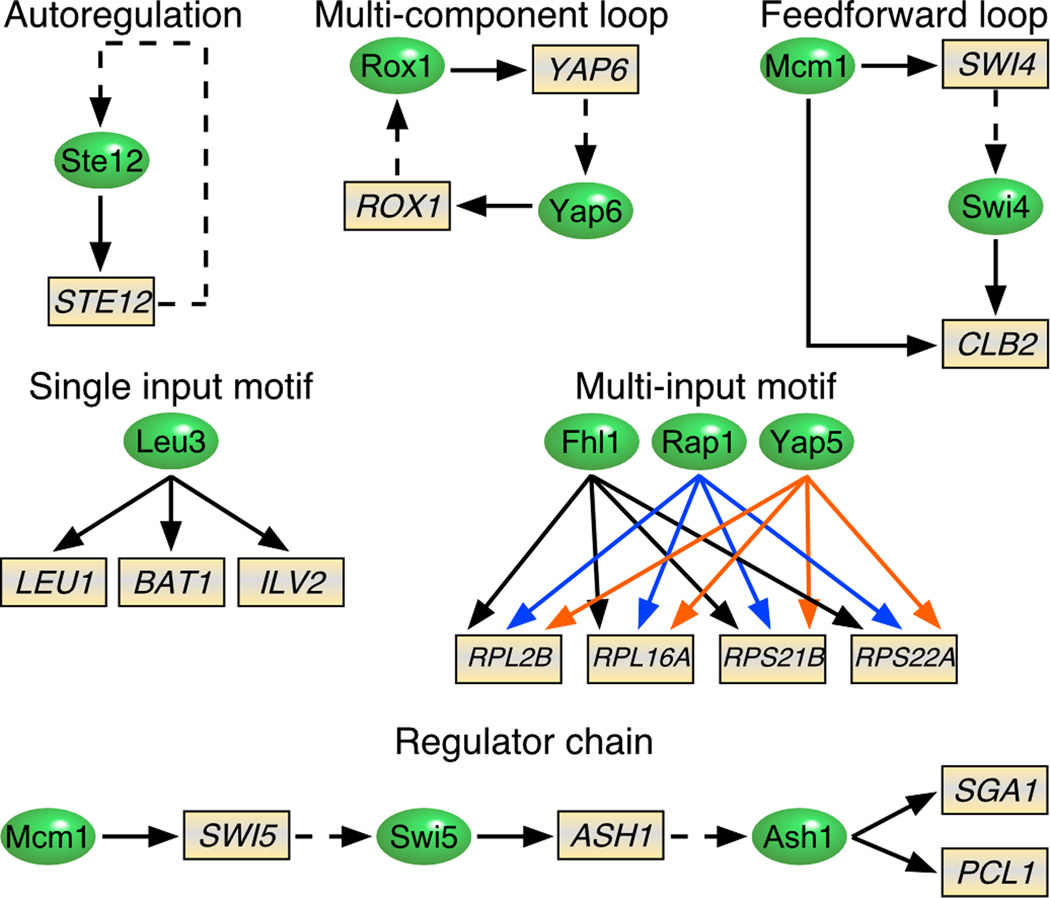
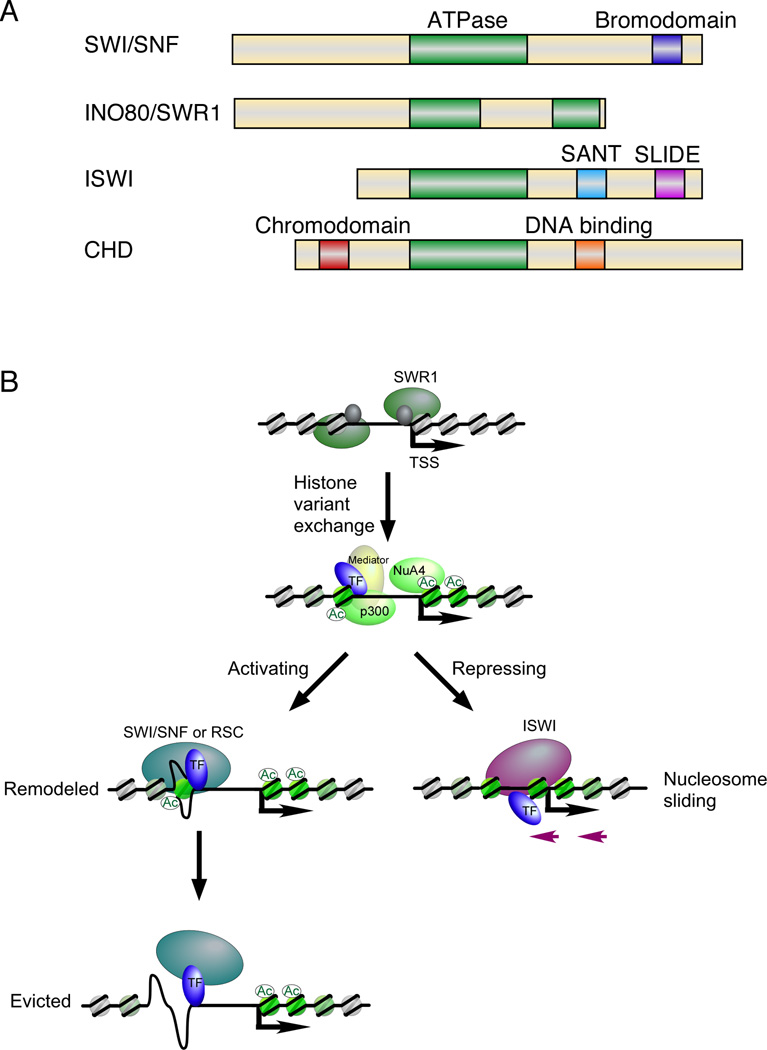
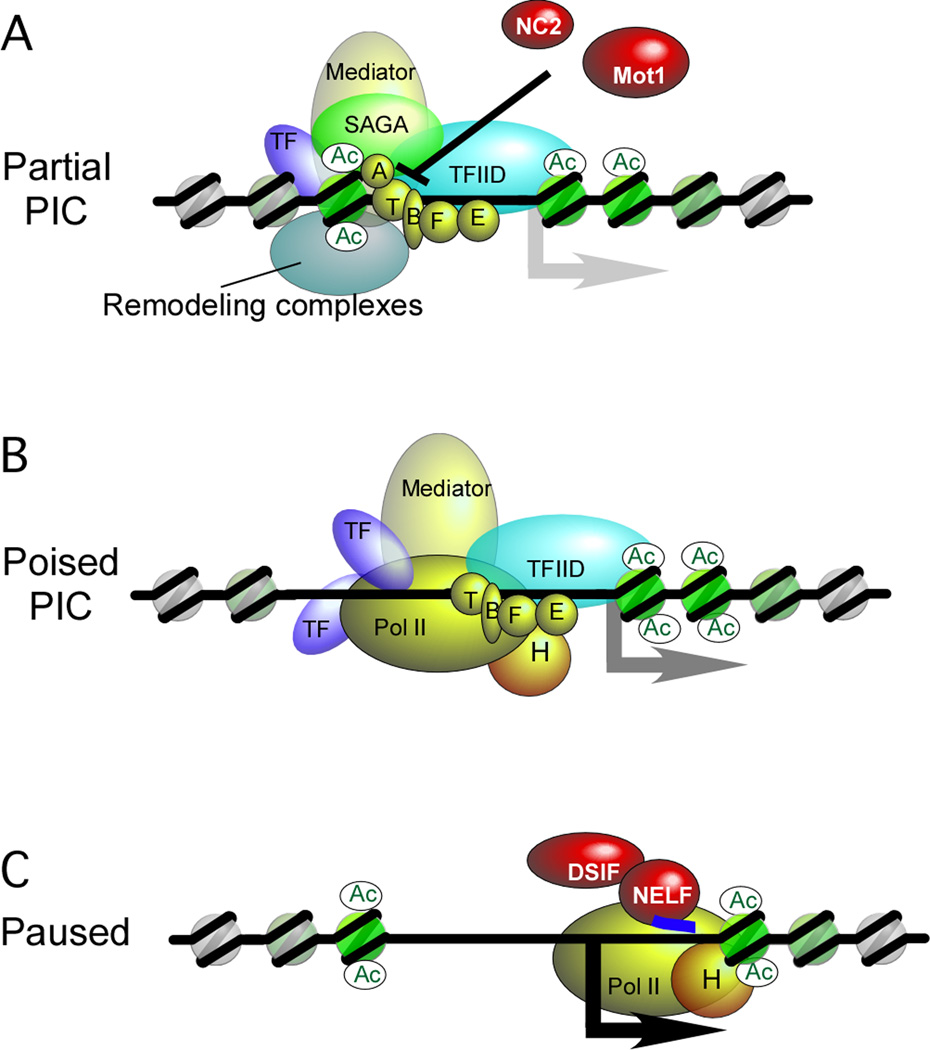
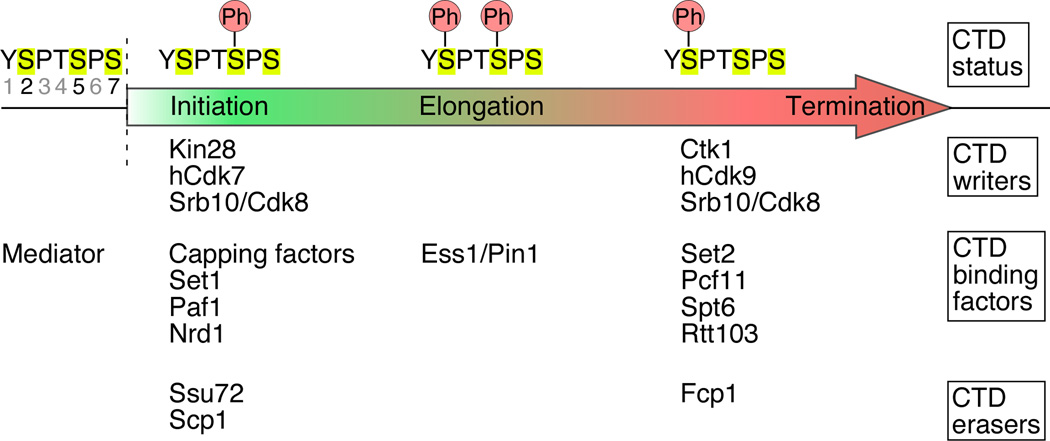
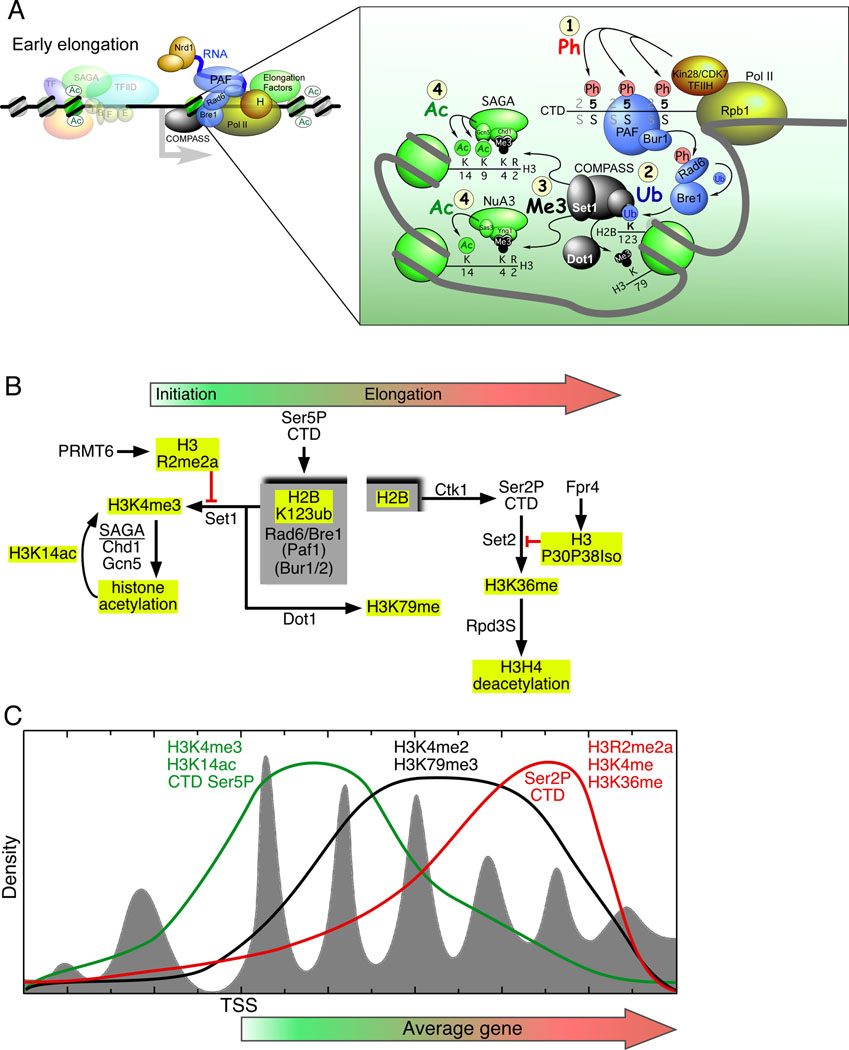
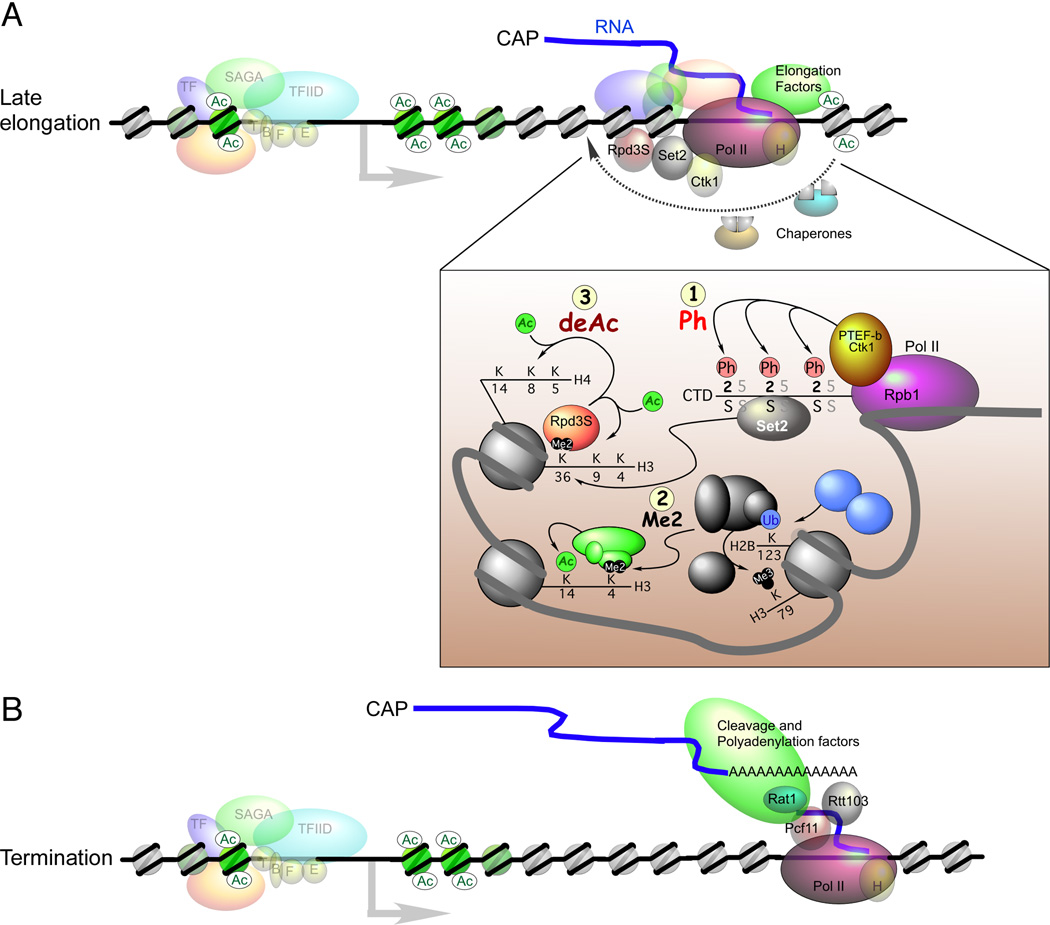
Regulation of eukaryotic gene expression is far more complex than one might have imagined 30 years ago. However, progress towards understanding gene regulatory mechanisms has been rapid and comprehensive, which has made the integration of detailed observations into broadly connected concepts a challenge. This review attempts to integrate the following concepts: (1) a well-defined organization of nucleosomes and modification states at most genes; (2) regulatory networks of sequence-specific transcription factors; (3) chromatin remodeling coupled to promoter assembly of the general transcription factors and RNA polymerase II; and (4) phosphorylation states of RNA polymerase II coupled to chromatin modification states during transcription. The wealth of new insights arising from the tools of biochemistry, genomics, cell biology, and genetics is providing a remarkable view into the mechanics of gene regulation.
Figures

References
-
- Ahn SH, Kim M, Buratowski S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3' end processing. Mol Cell. 2004;13:67–76. - PubMed
-
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. - PubMed
-
- Angus-Hill ML, Schlichter A, Roberts D, Erdjument-Bromage H, Tempst P, Cairns BR. A Rsc3/Rsc30 zinc cluster dimer reveals novel roles for the chromatin remodeler RSC in gene expression and cell cycle control. Mol Cell. 2001;7:741–751. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources