

Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa
- PMID: 19520207
- PMCID: PMC2694974
- DOI: 10.1016/j.ajhg.2009.05.007
Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa
Abstract
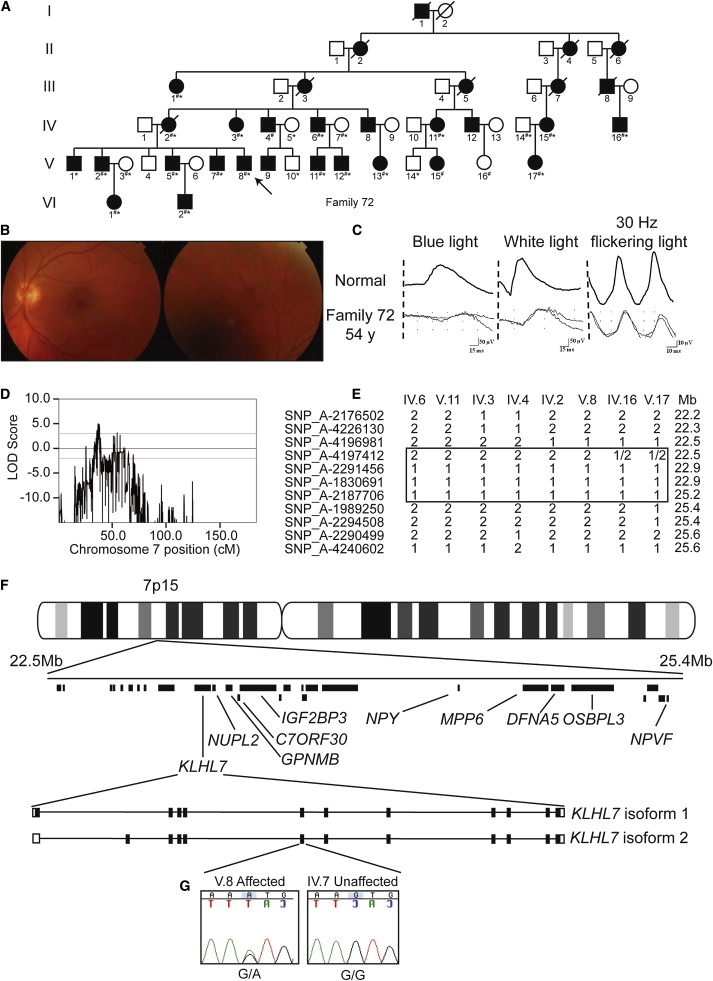
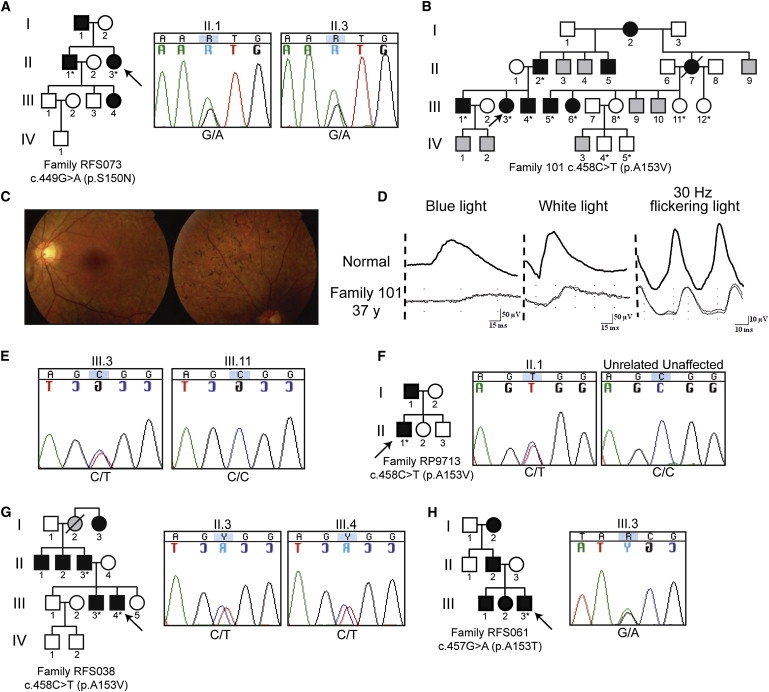
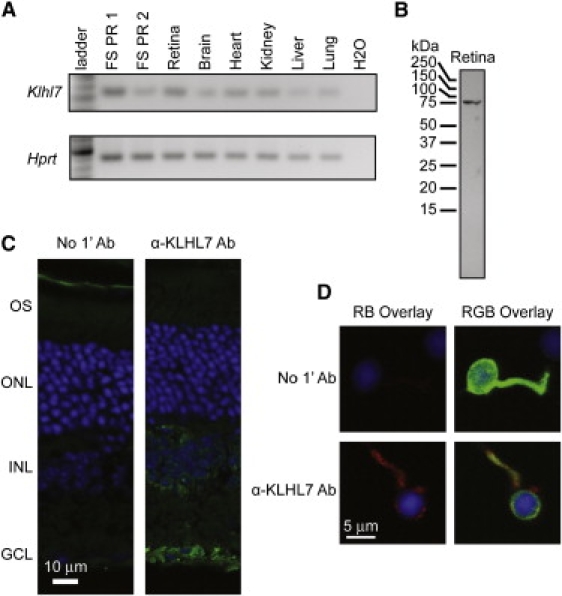
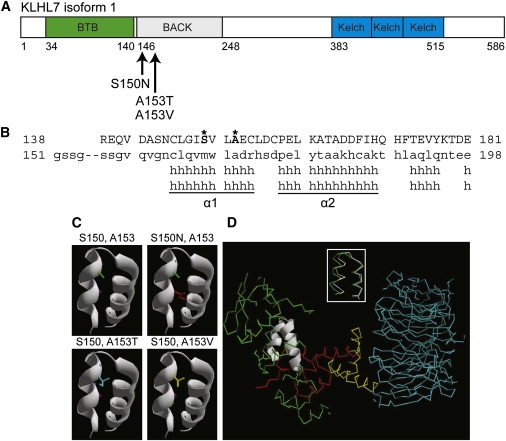
Retinitis pigmentosa (RP) refers to a genetically heterogeneous group of progressive neurodegenerative diseases that result in dysfunction and/or death of rod and cone photoreceptors in the retina. So far, 18 genes have been identified for autosomal-dominant (ad) RP. Here, we describe an adRP locus (RP42) at chromosome 7p15 through linkage analysis in a six-generation Scandinavian family and identify a disease-causing mutation, c.449G-->A (p.S150N), in exon 6 of the KLHL7 gene. Mutation screening of KLHL7 in 502 retinopathy probands has revealed three different missense mutations in six independent families. KLHL7 is widely expressed, including expression in rod photoreceptors, and encodes a 75 kDa protein of the BTB-Kelch subfamily within the BTB superfamily. BTB-Kelch proteins have been implicated in ubiquitination through Cullin E3 ligases. Notably, all three putative disease-causing KLHL7 mutations are within a conserved BACK domain; homology modeling suggests that mutant amino acid side chains can potentially fill the cleft between two helices, thereby affecting the ubiquitination complexes. Mutations in an identical region of another BTB-Kelch protein, gigaxonin, have previously been associated with giant axonal neuropathy. Our studies suggest an additional role of the ubiquitin-proteasome protein-degradation pathway in maintaining neuronal health and in disease.
Figures
References
-
- Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. - PubMed
-
- Sullivan L.S., Bowne S.J., Birch D.G., Hughbanks-Wheaton D., Heckenlively J.R., Lewis R.A., Garcia C.A., Ruiz R.S., Blanton S.H., Northrup H. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Invest. Ophthalmol. Vis. Sci. 2006;47:3052–3064. - PMC - PubMed
-
- Sancho-Pelluz J., Arango-Gonzalez B., Kustermann S., Romero F.J., van Veen T., Zrenner E., Ekstrom P., Paquet-Durand F. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol. Neurobiol. 2008;38:253–269. - PubMed
-
- Ruschendorf F., Nurnberg P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
