

Genome scan, fine-mapping, and candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype-specific differences in linkage and association results
- PMID: 19521098
- PMCID: PMC2709160
- DOI: 10.1159/000224636
Genome scan, fine-mapping, and candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype-specific differences in linkage and association results
Abstract
Objectives: Non-syndromic orofacial clefts, i.e. cleft lip (CL) and cleft palate (CP), are among the most common birth defects. The goal of this study was to identify genomic regions and genes for CL with or without CP (CL/P).
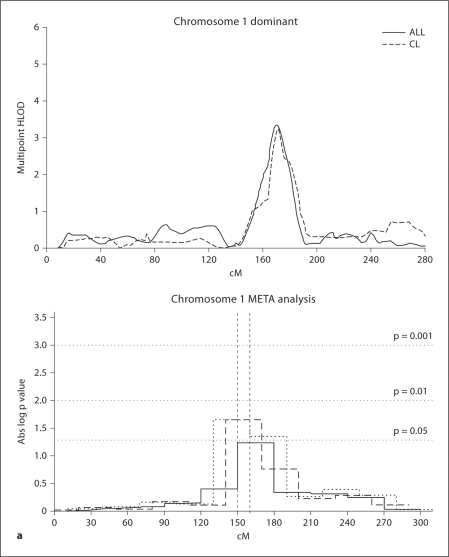
Methods: We performed linkage analyses of a 10 cM genome scan in 820 multiplex CL/P families (6,565 individuals). Significant linkage results were followed by association analyses of 1,476 SNPs in candidate genes and regions, utilizing a weighted false discovery rate (wFDR) approach to control for multiple testing and incorporate the genome scan results.
Results: Significant (multipoint HLOD >or=3.2) or genome-wide-significant (HLOD >or=4.02) linkage results were found for regions 1q32, 2p13, 3q27-28, 9q21, 12p11, 14q21-24 and 16q24. SNPs in IRF6 (1q32) and in or near FOXE1 (9q21) reached formal genome-wide wFDR-adjusted significance. Further, results were phenotype dependent in that the IRF6 region results were most significant for families in which affected individuals have CL alone, and the FOXE1 region results were most significant in families in which some or all of the affected individuals have CL with CP.
Conclusions: These results highlight the importance of careful phenotypic delineation in large samples of families for genetic analyses of complex, heterogeneous traits such as CL/P.
Copyright 2009 S. Karger AG, Basel.
Figures









References
-
- Trew CJ. Sistens plura exempla palati deficientis. Nova Acta Physio-Medica Academiae Caesareae Leopoldion-Carolinae. 1757;1:445–447.
-
- Darwin C. The Variation of Plants and Animals under Domestication. New York: Appleton; 1875.
-
- Sproule J. Hereditary nature of hare-lip. Br Med J. 1863;1:412.
-
- Rischbieth H. Hare-lip and cleft palate. In: Pearson K, editor. Treasure of Human Inheritance, part iv. London: Dulau; 1910. pp. 79–123.
Publication types
MeSH terms
Grants and funding
- N01 HG065403/HG/NHGRI NIH HHS/United States
- KO2-DE015291/DE/NIDCR NIH HHS/United States
- R01-DE012472/DE/NIDCR NIH HHS/United States
- R01 DE016148/DE/NIDCR NIH HHS/United States
- R01 DE012472/DE/NIDCR NIH HHS/United States
- R01-DE014677/DE/NIDCR NIH HHS/United States
- R01-DE09886/DE/NIDCR NIH HHS/United States
- R01 DE008559/DE/NIDCR NIH HHS/United States
- M01 RR000084/RR/NCRR NIH HHS/United States
- K02 DE015291/DE/NIDCR NIH HHS/United States
- R01 DE009886/DE/NIDCR NIH HHS/United States
- M01-RR00084/RR/NCRR NIH HHS/United States
- R03 DE016632/DE/NIDCR NIH HHS/United States
- R00 DE018085/DE/NIDCR NIH HHS/United States
- R01-DE016148/DE/NIDCR NIH HHS/United States
- R37 DE008559/DE/NIDCR NIH HHS/United States
- R03-DE016632/DE/NIDCR NIH HHS/United States
- R01 DE014667/DE/NIDCR NIH HHS/United States
- R37-DE08559/DE/NIDCR NIH HHS/United States
- P50 DE016215/DE/NIDCR NIH HHS/United States
- P50-DE016215/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
